
Tom Goddard
February 11, 2010
AMI Forum, Scripps
|
|
| Greg Pintilie developer of Segger |
Show groel and bacteriophage lambda examples from Segger documentation. Groel EMD 1080 (Steve Ludtke) and monomer 1xck, chain A. Segment Open emd_1080.map Show Segger dialog. Warn that it is an odd user interface, 8 menus, 3 columns. Press segment button. Set white background and silhouette edges and glossy lighting (shortcuts wb, se, gl). Explain each region is associated one local maximum, including all grid points that reach that local maximum after travelling a steepest ascent point. Mention that only parts of map above displayed threshold used. Group Goal is to group these regions to form larger regions each representing one molecule. Groel has 14 copies of one molecule. That is done by smoothing map, giving fewer local maxima, and segmenting smoothed map, then use those regions to group all the original map regions that overlap a single smoothed map region. That is done iteratively. Set smoothing steps to 5, step size 10, stop at 14 regions. Step size is Gaussian standard deviation (I checked code). Press smooth and group button. Note that the iterative smoothing is stopped before the requested number of steps if we get down to 14 regions. Actually takes only 3 steps as seen in comments on the status line. Ungroup Select a region ungroup it, select an ungrouped region and ungroup again. Select all regions of original selection and group. Mask Show mask map with selected. Fit molecule Fit groel monomer, open 1xck_A.pdb (chain A of 1xck). Choose 1xck_A.pdb in segger dialog. Select a region to fit to (different from ungrouped/regrouped one to avoid bug where fit goes to wrong region). Note that fit will use a simulated map with specified resolution, grid spacing. Use Align to "selected region". Explain that this alignment took the principal axes of the molecule based on inertia and aligned them to the principal axes of the region density, then locally optimized with a rigid rotation and translation using Chimera's usual fit optimization routine. Principal axes Show molecule axes and region axes. Note that axes could be anti-parallel. Segger tries 4 unique combinations (all parallel, or any two anti-parallel), locally optimizes each, and takes highest correlation. Note that axes not perfectly aligned because of local optimization. Saving fit Show save fitted structure menu entry. Saves PDB coordinates that will properly align with the map. Global rotation fit Show fit by rotation menu entry that tries local optimization from 64 uniformly distributed starting orientations with center of mass of molecule and region matching. Might show to show how fast it is. It moves principal axis fit only slightly. Principal axes fit will be unreliable for spherical shaped molecule.Bacteriophage lambda example
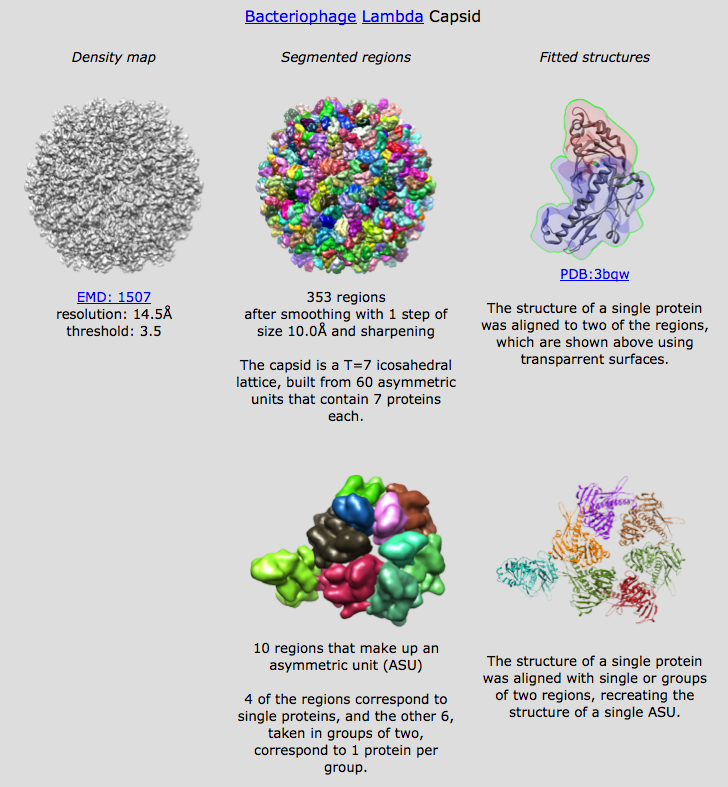
Segment and Group Open emd_1507.map, threshold to avoid most dust (2.71), set minimum region size to 100 voxels, press Segment Use 1 smoothing step, size 10, press Smooth and Group Asymmetric unit T=7 virus capsid, expect about 7*60/2 = 210 copies of capsid protein. Find and show pentamer looks like regions are whole molecules. Hexamers have some molecules represented by 2 regions. Show asymmetric unit. Select each region of hexamer and one neighbor pentamer. Use "show only selected". Inspect bottom of hexamer for missing pieces. (Intentionally miss one in initial selection). Add missing piece to selection. Show all, Invert selection and delete and delete selected regions. More smoothing creates regions that are bigger than one molecule. Could manually group the regions but let's imagine we weren't sure of the proper groupings. Fit to pentamer Fit pdb 3bqw, a putative E. coli prophage capsid protein. A prophage is a virus genome incorporated into a bacterial host genome. This one has 43% identical sequence. Select pentamer region and fit 3bqw. Good fit except for a beta sheet. Fit to union of 2 regions Select two regions of hexamer that for a monomer and align to "combined selected regions". Fits nicely. Fit to calculated region groups Try align to "groups of regions". This takes a minute or two, so start it then explain. Adjacent regions are combined so their volume is at least as big as the volume of the fit molecule but not too big -- removing any region from a group would make it smaller. Further filter out groups that are more than 50% bigger than predicted or differ by more than 10% in bounding radius. I get 25 groups (threshold 2.71), see them counted off in status line. Grouping works by joining adjacent regions to create regions whose volume is between 25% and 175% of target volume determined as volume enclosed in simulated map. Radius has to be +/- 20% of target map radius from center to most distant boundary point. Place fits for full asymmetric unit Use Place button to place 7 copies in top scoring fits. Show text file which shows other fits. Need dialog user interface to make it easier to browse those fits by say clicking an entry in a list.