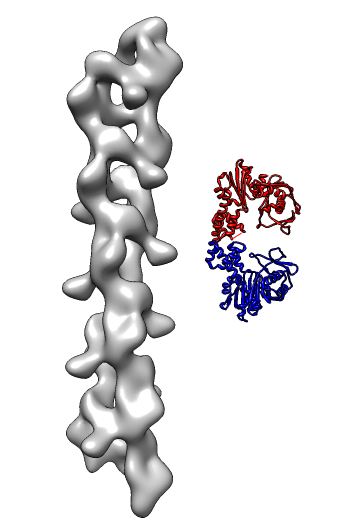
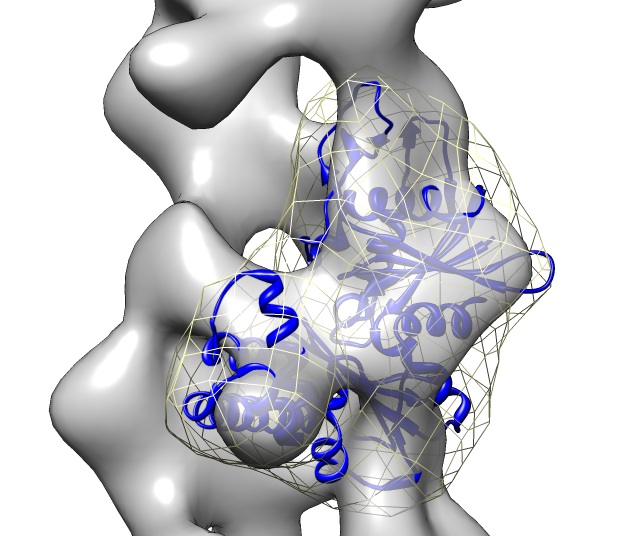
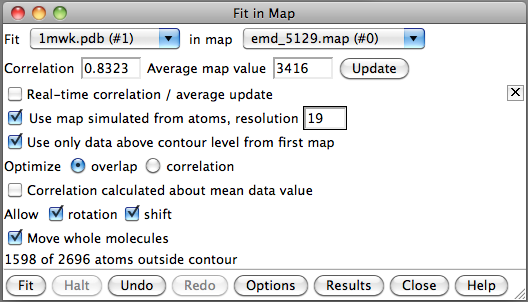
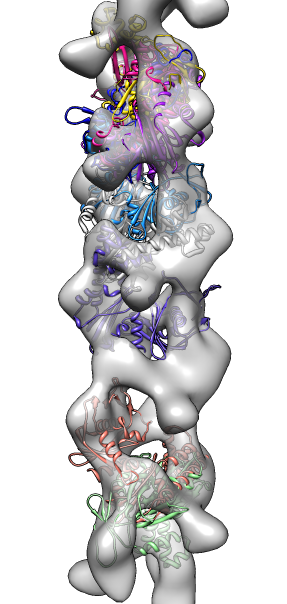
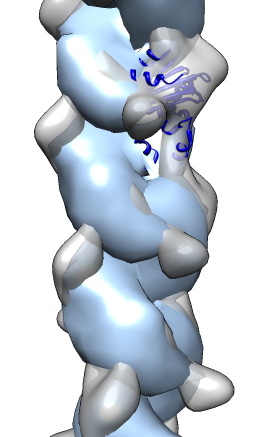
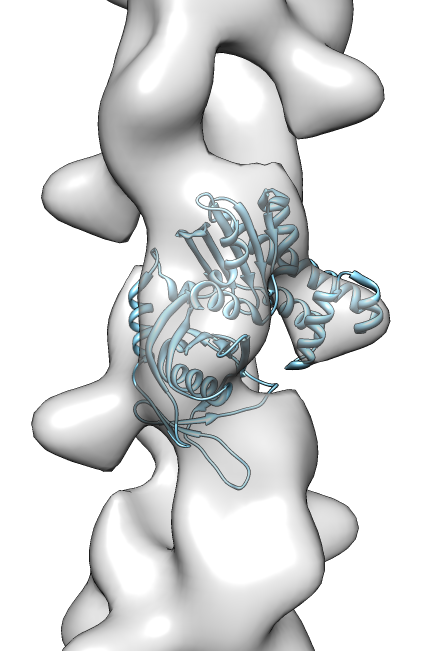
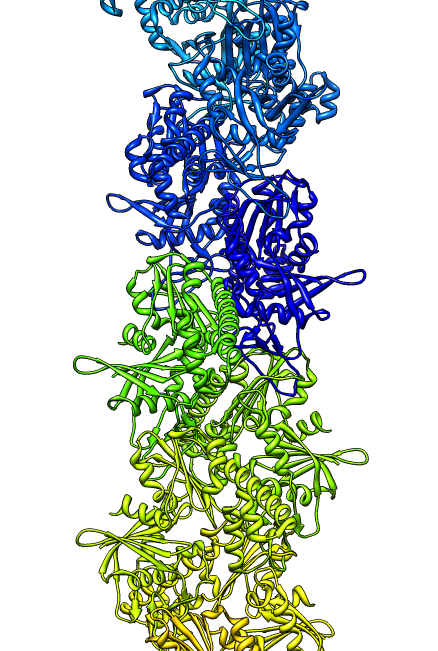
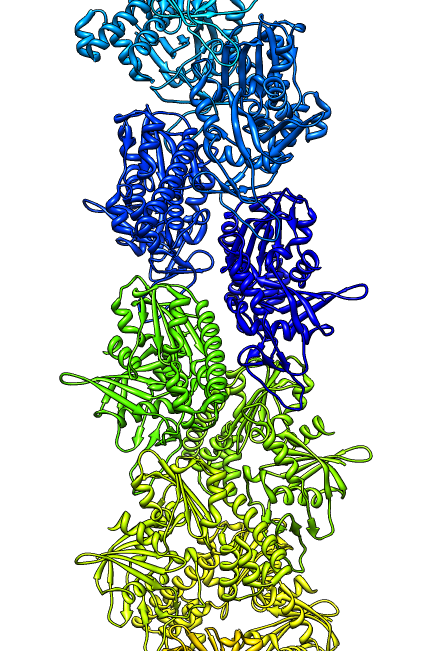
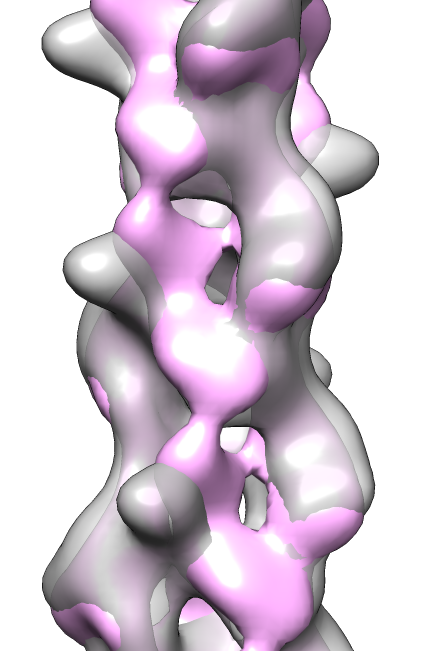
- Search for best fit using 30 tries with command "fit #1 #0 search 30"
- Fits appear all along filament, many are equivalent due to symmetry
of the filament.
|
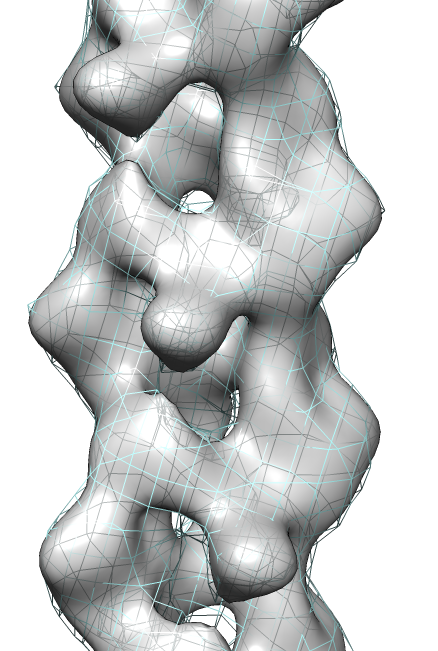
- Determine map symmetry to eliminate equivalent fits.
- Command "measure symmetry #0 helix 20,180,opt minimumCorrelation 0.95".
- The "helix" option gives Chimera a hint about helical parameters.
- The "minimumCorrelation" option accounts for this unusual map where
the helix does not extend to the edges of the volume box.
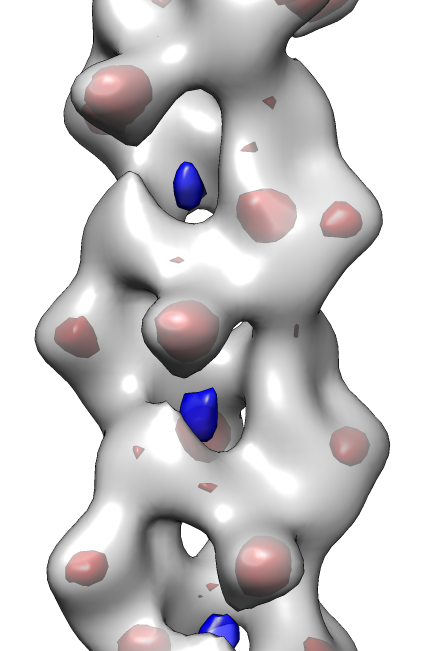
- View symmetry copies of molecule with command "sym #1 group #0 surf true".
- Remove symmetry copies with "~sym #1" (note leading tilde character
which means "undo" in Chimera commands).
|
- Clear fit list.
- Rerun previous fitting command "fit #1 #0 search 50".
- Clear fit list.
- Use correlation optimization "fit #1 #0 search 50 res 19".
|