Chimera-Related Databases and Software
See also:
Web services used by UCSF Chimera,
sites that provide
Chimera web data,
Chimera
Python scripts
ASTRAL
Experimentally determined protein structures, divided into domains
as defined by SCOP
(Structural Classification of Proteins).
Cancer3D
 |
Database of cancer missense mutations in the context of 3D structures,
disease classifications, and patient characteristics.
The 3DMol window for viewing associated PDB structures
includes an option to download a Chimera session of the structure
with mutated positions shown and colored by mutation frequency.
The Cancer3D database is described in
Sedova et al.,
Nucleic Acids Res 47:D895 (2019).
|
EM DataBank (EMDB)
|
This site contains 3D density maps obtained by electron microscopy in
CCP4 format that can be shown with Chimera
Volume Viewer.
|
EM Navigator
Presents images, movies, Chimera sessions and details about electron microscopy
density maps from the EMDB.
ModBase
A database of three-dimensional protein models calculated by
comparative modeling.
The website includes links for viewing models in Chimera
along with the corresponding template structure and template-target
sequence alignment, or colored to show binding site predictions.
ModBase models and associated information can also be
fetched
directly into Chimera.
Mutagenetix
A database of mouse mutations and phenotypes induced by ENU.
Many of the allele pages include molecular structure images and
movies made in Chimera.
PhosphoSitePlus®
 |
Comprehensive information and tools for the study of protein
post-translational modifications; includes scripts for visualization
in Chimera.
|
RCSB Protein Data Bank (PDB),
Protein Data Bank in Europe (PDBe),
Protein Data Bank Japan (PDBj)
Experimentally determined protein and nucleic acid structures.
Display PDB and mmCIF format files of atomic coordinates.
UCSC Genome Browser
Chimera links are provided in UCSC Gene pages
(see example) and SNP track details pages.
Clicking a Chimera link displays the corresponding structure
with nonsynonymous SNP residues colored and
labeled with the dbSNP identifier.
Virus Particle Explorer

|
Atomic resolution virus capsid structures. Some of the capsid images on
this site are made with the Chimera
Multiscale Models
tool.
|
3DFSC Program Suite
Software for 3D Fourier shell correlation analysis of cryoEM data,
available from GitHub.
The program requires Anaconda to run, and uses Chimera to display some outputs.
See
Tan et al., Nat Methods 14(8):793 (2017).
3V: Voss Volume Voxelator
AlphaSpace
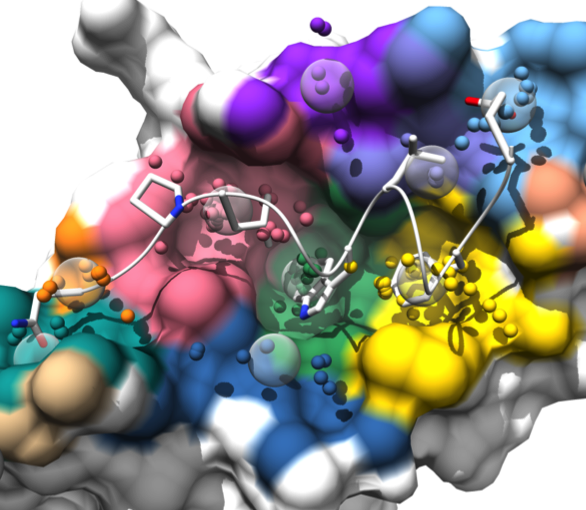 |
AlphaSpace is a tool for analyzing surface pockets
at protein-protein interfaces.
AlphaSpace generates a Chimera session file with several “scenes”
for viewing the results in different ways.
The program is available upon request from the authors for academic use
and is described in Rooklin et al.,
J Chem Inf Model 55:1585 (2015).
|
Amber
Molecular force field and dynamics simulations.
Display trajectories with the Chimera
MD Movie extension.
Box Beam Backbone Sculptures
CanDo
Atomic Model Generator
 |
Uses Chimera to generate atomic models of DNA nanostructures from the
description in the CanDo (.cndo) file format. Chimera is also used to
generate
3D printable models from CanDo.
|
CoCoPOD
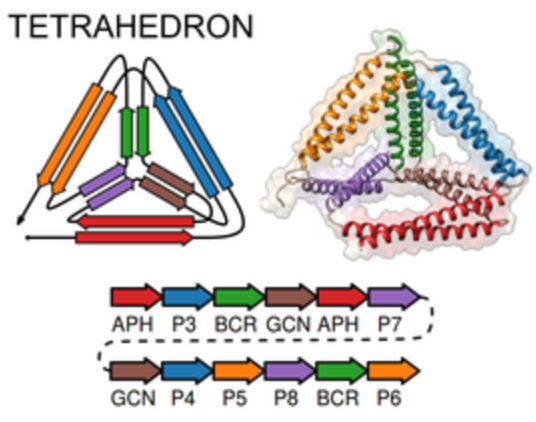 |
The CoCoPOD platform for Coiled-Coil Protein Origami Design is
available on GitHub and described in
Ljubetič et al.,
Nat Biotechnol 35:1094 (2017).
Dependencies of the software include Chimera.
|
ConSurf Server
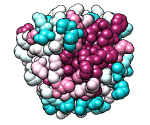 |
Given a query structure, the server finds related sequences, aligns them,
and displays conservation on the structure.
Clicking the link to view results in Chimera uses the
Chimera web data mechanism to display the structure and
sequence alignment colored by conservation, as well as the
ConSurf-calculated phylogenetic tree and custom alignment headers
(example).
|
Direct Coupling Analysis
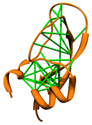 |
Direct Coupling Analysis (DCA) is a statistical inference framework
for identifying direct co-evolutionary couplings among residue pairs
in multiple sequence alignments. A protein multiple sequence alignment can be
uploaded to the web service, or the code can be downloaded for local use.
The download includes a Matlab implementation
and scripts for 3D visualization in Chimera as described in
Morcos et al.,
Methods Mol Biol 1137:55 (2014).
|
DOCK
Given 3D structures of molecules, DOCK addresses the problem of how they
might fit or bind together. Using Chimera, one can:
- prepare structures as input to DOCK (or other programs) with
Dock Prep
- save a
DMS
dot molecular surface (used for DOCK sphere generation)
- screen DOCK results with the
ViewDock extension
- view scoring grids with
Volume Viewer
DROIDS
DROIDS is a pipeline for GPU-accelerated comparative protein dynamics,
incorporating graphical user interface control for Amber MD,
cpptraj analysis, and statistical and visual representations
in R graphics and UCSF Chimera. DROIDS is described in
Babbitt et al., Biophys J 114:1009
(2018), and open-source code is
available on GitHub.
EMAN2 –
Electron Micrograph Analysis
|
Image processing suite for single-particle reconstruction from cryo-EM
and working with data from related subdisciplines
such as tomography and 2D crystallography.
The previous program EMAN1 included the
EMANimator extension
for making volume-data (map) animations in Chimera.
|
JEvTrace
JEvTrace is a Java implementation of the evolutionary trace method.
Results can be shown in Chimera as coloring of the sequence alignment
and any associated structures by reading a
JEvTrace output file into
Multalign Viewer.
Kalasanty:
Binding Site Prediction
Kalasanty uses a neural network to predict binding sites in protein structures.
The predictions can be saved as .cmap or .cube grid files for display
in Chimera or other programs. Kalasanty is described in
Stepniewska-Dziubinska et al.,
Sci Rep 10:5035 (2020) and is available as
source code.
LabelHash Server
The server searches the PDB for matches to a user-specified
3D motif (pattern of residues). The authors provide the Chimera extension
ViewMatch
for looking through the hits superimposed on the query.
LabelHash can also be
downloaded
for local command-line use.
MacroMoleculeBuilder (MMB)
MMB is a multiresolution modeling tool for building 3D structural
and dynamical models of macromolecules, with explicit control
over degrees of freedom, forces, and constraints;
it was previously known as RNABuilder but now also handles protein and DNA.
MMB is available for
download
along with a plugin for using Chimera as the GUI, as described in
Tek
et al., Nucleic Acids Res 44:95 (2016).
MOLEonline Server
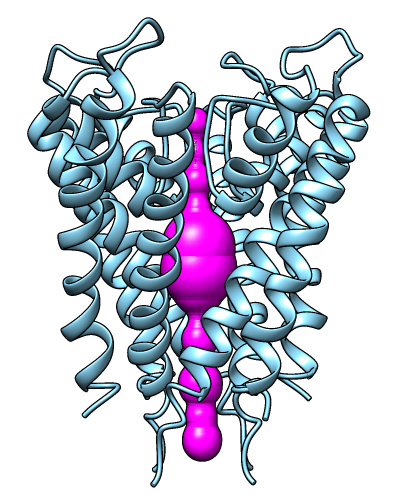 |
The MOLEonline server runs MOLE, a program
for identifying and characterizing channels in macromolecular structures.
Results include a profile of the channel radius and hydropathy,
a list of residues lining the channel, and scripts for display
in Chimera and other programs. The server is described in
Pravda et al., Nucleic Acids Res
46:W368 (2018).
The image shows results for PDB 1BL8 biological unit calculated in pore mode,
displayed in Chimera.
See also: bash script
for visualizing HOLE
output in Chimera, as described in associated posts:
[011372]
[011374]
|
PDBmutator
A GUI-enabled script to batch-run Chimera virtual mutation commands
(swapna or swapaa) on a PDB file to create all possible structural mutants
or all variants defined by a CLUSTAL protein sequence alignment.
Partial Order Structure Alignment
(POSA) Server
 |
The POSA server performs flexible multiple structure alignment.
The results visualization page includes a link to download
a python script for viewing the superimposed structures in Chimera.
The POSA server is described in
Li et al.,
Nucleic Acids Res 42:W240 (2014).
|
PyChimera
PyChimera
provides access to the UCSF Chimera codebase from any Python 2.7 interpreter,
making it easier to integrate with other software.
PyChimera is described in
Rodríguez-Guerra Pedregal and Maréchal,
Bioinformatics bty021 (2018).
PyTom
Unifies standard tomogram processing steps in a single python-based toolbox;
modified version of Chimera's Volume Viewer available from the
PyTom website.
ResMap
ResMap (Resolution Map) is a Python application for computing the
local resolution of cryo-EM density maps. It generates a map of
the local resolution and a Chimera script for 3D visualization.
ResMap is available
on SourceForge and described in
Kucukelbir et al.,
Nature Methods 11:63 (2014).
See also: LocalFSC
Ringer
 |
The Ringer program detects molecular motions by systematically sampling
X-ray electron density; the aim is to go beyond static structural snapshots
of proteins by uncovering structural ensembles.
The program depends on Chimera.
Ringer is available for
download
and is described in
Lang
et al., Protein Sci 19:1420 (2010).
|
ScanNet Server
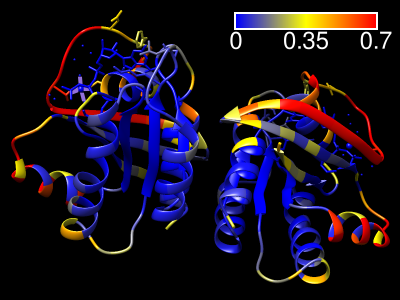 |
The ScanNet server predicts protein-binding sites for
structures in the PDB, the AlphaFold Database, or uploaded from a local file.
The method is described in
Tubiana, Schneidman-Duhovny, and Wolfson,
Nat Methods 19:730 (2022)
and the web server specifically in
Tubiana et al.,
J Mol Biol 434:167758 (2022).
Results are emailed to the user as a zip file containing:
- the PDB file with binding site probabilities in the B-factor field
- csv file containing the binding site probabilities
- ChimeraX command script (.cxc) for visualizing the results
(right-click → open the file with ChimeraX)
- Chimera python script (.py) for visualizing the results
(right-click → open the file with Chimera)
|
SketchBio
A tool for molecular modeling and animation
that uses Chimera for surface generation.
Sparky
Assignment of NMR spectra for determining protein and nucleic acid structures.
Uses Chimera to
display NMR peak assignments
on a molecular model.
structureViz2
 |
A Cytoscape app
that integrates protein networks or pathways in Cytoscape
with visualization and analysis of corresponding structures in Chimera.
It also works with
RINalyzer to calculate
residue interaction networks (RINs) for Cytoscape from
protein structures in Chimera.
|
TALOS:
Three-dimensional, Algorithmically generated Library
of DNA Origami Shapes
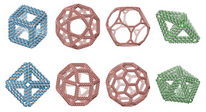 |
TALOS
is an open-source software package for converting
3D computer-generated design files (PLY) into DNA sequences
that can be synthesized and mixed to generate
DNA 6HB-based wireframe 3D nanoparticles with high fidelity.
TALOS also writes
BILD files
for display in Chimera.
|
ViewMotions Server
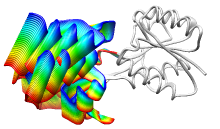 |
Given two protein conformations or
an ID from the Database of Macromolecular Movements, the server
generates a ViewMotions rainbow image and corresponding Chimera session.
The conformational change is shown as a series of interpolated
structures with variable regions rainbow-colored from blue to red.
The server is described in
Cockrell
and Kantrowitz, J Mol Graph Model 40:48 (2013).
|