Using AlphaFold protein structures in ChimeraX for cryoEM modeling
Tom Goddard
November 9, 2021
SBGrid / University of Otago Webinar Series
Presentation video.
ChimeraX AlphaFold capabilities
- Search for best sequence match in EBI AlphaFold database
- BLAST to get list of sequence matches in EBI AlphaFold database
- Predict a structure from a sequence.
Finding a Starting Atomic Model for a cryoEM Map
We try to find an initial atomic model for the human TACAN dimer structure
using the AlphaFold database at the EBI
and ChimeraX. This map and an atomic model were published August 2021,
had no prior know homologous models in the Protein Databank
and is thought to be a mechano-sensitive ion channel involved
in pain sensation or lipid metabolism enzyme.
Cryo-EM structures of human TMEM120A and TMEM120B.
Ke M, Yu Y, Zhao C, Lai S, Su Q, Yuan W, Yang L, Deng D, Wu K, Zeng W, Geng J, Wu J, Yan Z.
Cell Discov. 2021 Aug 31;7(1):77. doi: 10.1038/s41421-021-00319-5. PMID: 34465718.
The long intracellular alpha helix at the bottom can be rigidly moved with the ChimeraX
move atoms mouse mode to better fit the density to improve the initial model. Then
the atomic model can be refined in the map to correct side positions, e.g. with the ChimeraX
ISOLDE tool.
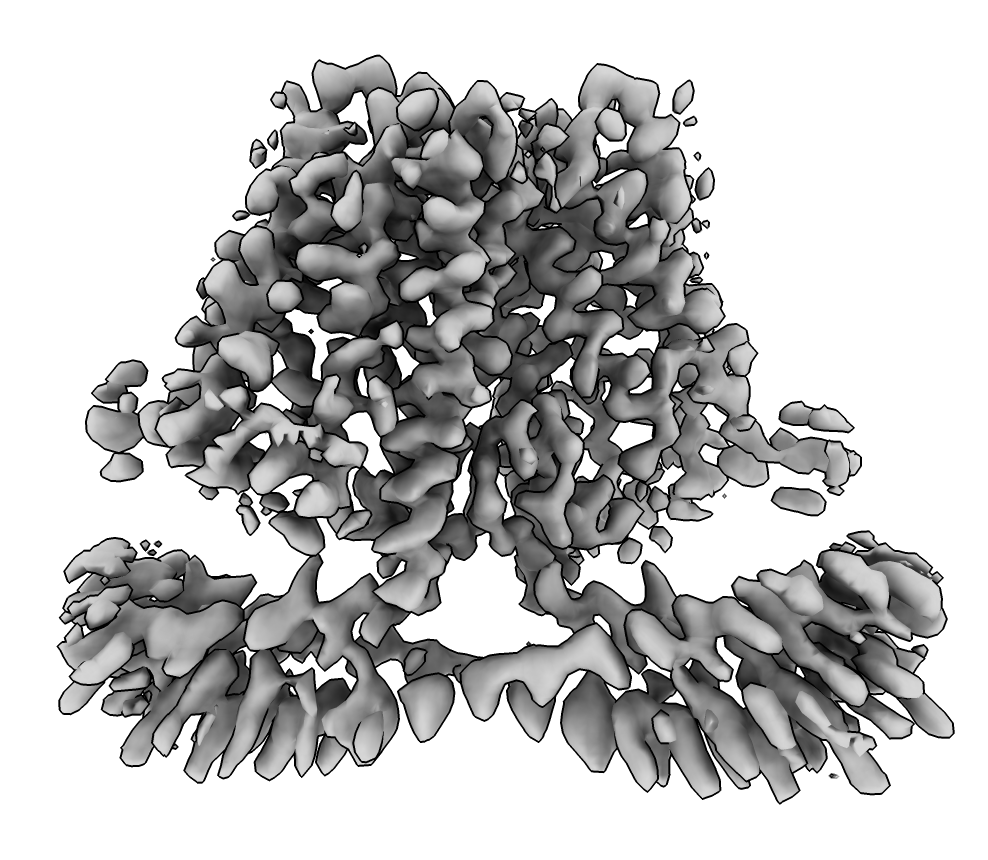
EMDB map 30495, 3.4 Angstroms.
(fetched with ChimeraX command open 30495 from emdb).
| 
ChimeraX 1.3 AlphaFold tool, in menu Tools / Structure Prediction,
with UniProt sequence TACAN_HUMAN,
then press Fetch button.
| 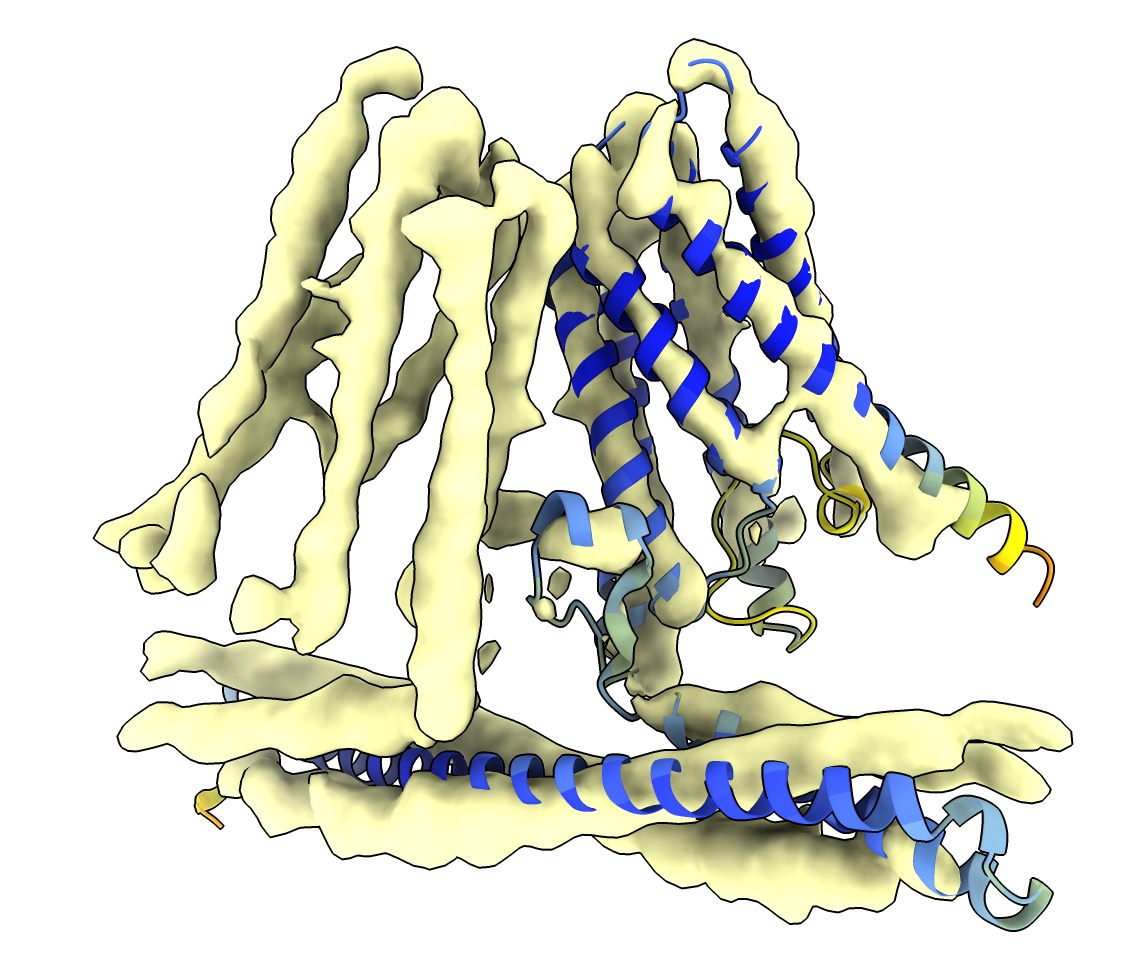
AlphaFold EBI database model fit into map (smoothed with volume gaussian #1 sdev 2).
|
Searching for AlphaFold Models
Now consider another example, a chicken membrane protein that transports omega-3
fatty acids, that is not in the AlphaFold EBI database which includes only
21 organisms. UniProt sequence
F1NCD6_CHICK.
Structural basis of omega-3 fatty acid transport across the blood-brain barrier.
Cater RJ, Chua GL, Erramilli SK, Keener JE, Choy BC, Tokarz P, Chin CF, Quek DQY, Kloss B,
Pepe JG, Parisi G, Wong BH, Clarke OB, Marty MT, Kossiakoff AA, Khelashvili G, Silver DL, Mancia F.
Nature. 2021 Jul;595(7866):315-319. doi: 10.1038/s41586-021-03650-9.
Closest sequence matches in AlphaFold database are from rat, human, zebrafish and mouse and are
about 70% identical.
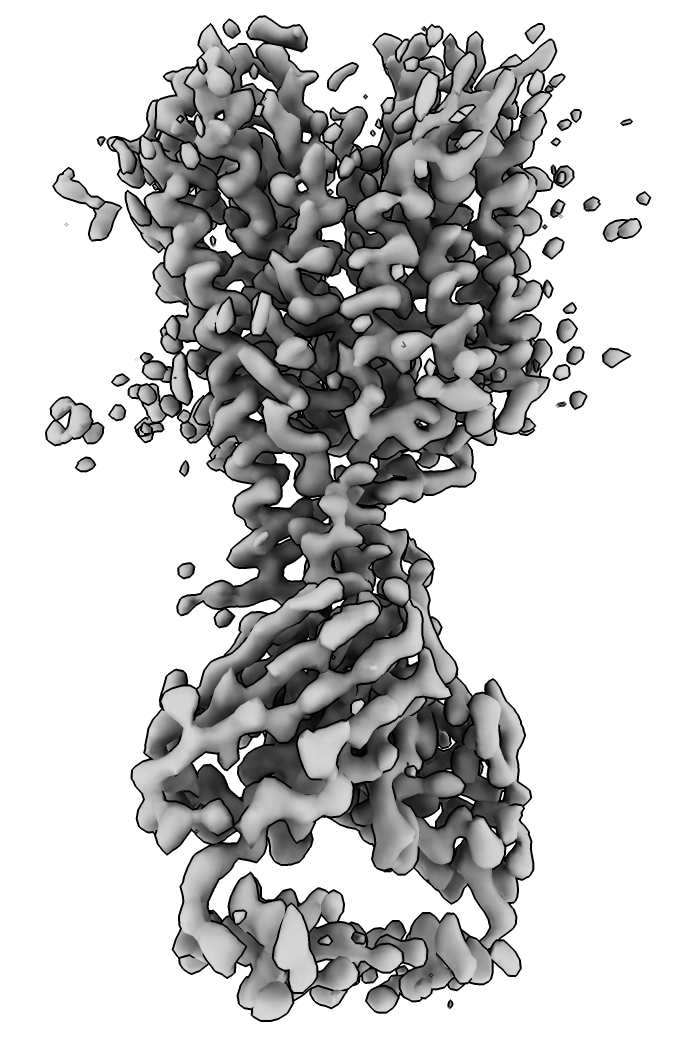
EMDB map 23883. Membrane protein top, antibody at bottom.
| 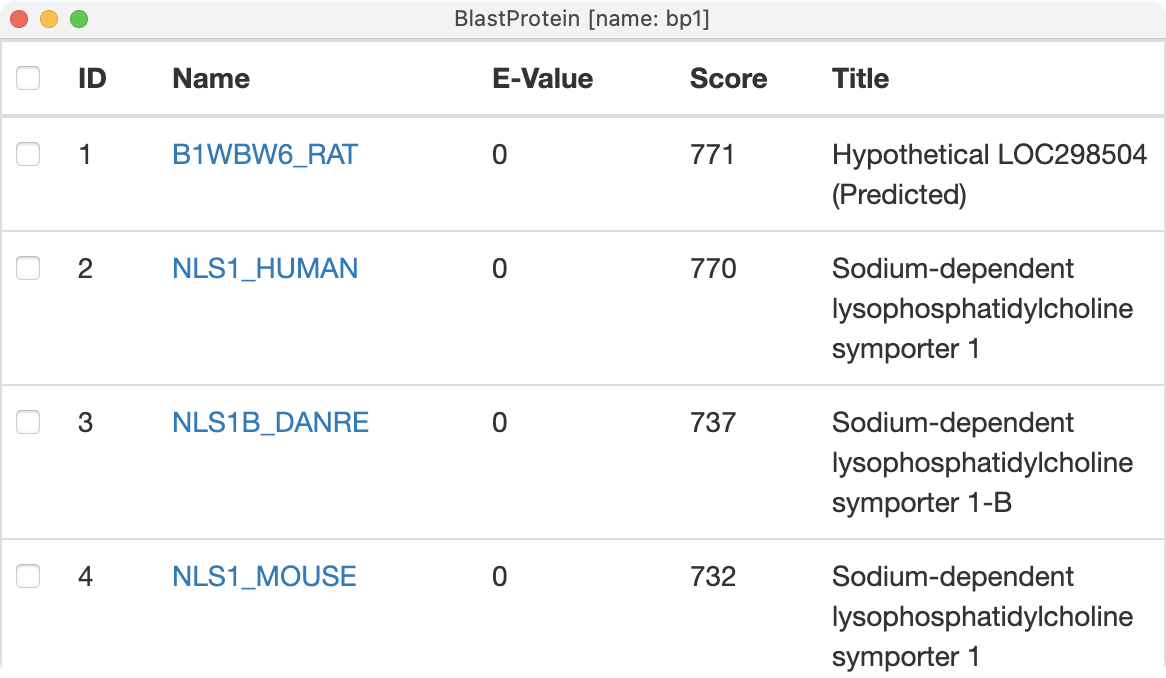
AlphaFold database BLAST search results.

Sequence similarity of closest match (rat).
| 
Four closest AlphaFold models
rat,
human,
zebrafish,
mouse.
|
Running AlphaFold to Predict a Structure from a Sequence
To predict the chicken sequence structure run AlphaFold by pressing the
Predict button on the
ChimeraX AlphaFold tool.
This will run
AlphaFold on Google Colab free servers. You will be asked to sign in
to your Google account (same account used for Google email, drive, calendar).
A security warning will display saying the ChimeraX AlphaFold code being
run is not from Google, click Run Anyway.
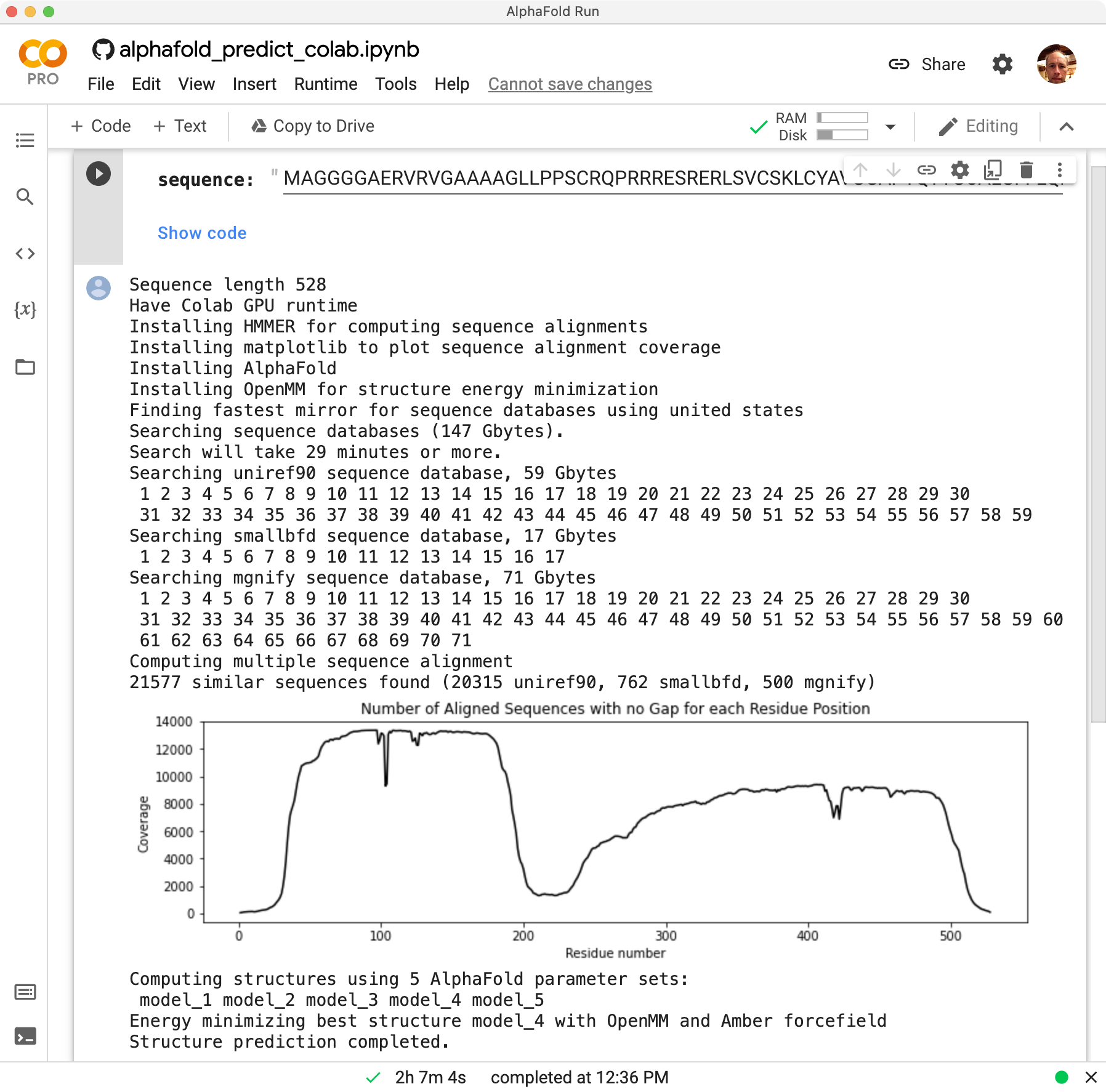
Output
The run took 2 hours 7 minutes to predict the 528 amino acid sequence.
Log output shows that it installed HMMER
(for computing a multiple sequence alignment),
AlphaFold,
and OpenMM
(to energy minimize final
structure), then searched 150 Gbytes of sequence databases
(uniref90, smallbfd, mgnify), then ran the AlphaFold neural net
with 5 alternative sets of parameters and selected the most confident
resulting structure to energy minimize. The structure then was
automatically loaded in ChimeraX. The
best model is downloaded to
your Downloads folder where ChimeraX keeps fetched files.
~/Downloads/ChimeraX/AlphaFold/prediction_29
671579 best_model.pdb
264500 mgnify_alignment
528003 mgnify_deletions
6 model_1_score
333851 model_1_unrelaxed.pdb
6 model_2_score
333851 model_2_unrelaxed.pdb
6 model_3_score
333851 model_3_unrelaxed.pdb
671579 model_4_relaxed.pdb
6 model_4_score
333851 model_4_unrelaxed.pdb
6 model_5_score
333851 model_5_unrelaxed.pdb
403098 smallbfd_alignment
804675 smallbfd_deletions
535 target.fasta
10746635 uniref90_alignment
21452771 uniref90_deletions
How does AlphaFold work?
- It uses sequence databases to make a deep multiple sequence alignment.
The databases have more than a billion sequences, basically all experimentally known
protein sequences. It typically makes a sequence alignment containing thousands of
sequences, sometimes over 100,000 sequences. If the alignment has fewer than 30 sequences
prediction quality can be bad. AlphaFold infers which residues contact each other
from residue covariation observed in the sequence alignment. That is how it figures
out the fold.
- AlphaFold can optionally use structure templates (from the Protein Databank).
These also help AlphaFold know the correct fold. The ChimeraX prediction does not
use structure templates.
- Uses multiple sequence alignnment and templates to predict distances between
every pair of residues, then constructs a structure using that residue pair distance map.
- Part of AlphaFold was trained to pack residues based on all known experimental
structures and the resulting residue packing is often very accurate.
Limitations of AlphaFold
- EBI AlphaFold database has only 21 organisms, human, mouse, zebrafish, arabidopsis, E. coli...
- AlphaFold predictions are just for single proteins not complexes.
- Predicting one structures takes 1 to 20 hours depending on sequence length.
- AlphaFold fails for longer sequences 800 - 2500 amino acids depending on amount of available GPU memory.
- AlphaFold does not handle ligands, ions, solvent.
- Running AlphaFold requires a modern high-end Nvidia GPU (uses CUDA) and Linux.
- AlphaFold uses large databases, 2 Tbytes, that can take days to download.
AlphaFold EBI Database Species
The EBI AlphaFold database has predictions for 21 organisms.
From the EBI database:
"In the coming months we plan to expand the database to cover a large proportion of all catalogued proteins (the over 100 million in UniRef90)."
| Common Name | Predicted
Structures | Species
|
|---|
| Arabidopsis | 27,434 | Arabidopsis thaliana
|
| Nematode worm | 19,694 | Caenorhabditis elegans
|
| C. albicans | 5,974 | Candida albicans
|
| Zebrafish | 24,664 | Danio rerio
|
| Dictyostelium | 12,622 | Dictyostelium discoideum
|
| Fruit fly | 13,458 | Drosophila melanogaster
|
| E. coli | 4,363 | Escherichia coli
|
| Soybean | 55,799 | Glycine max
|
| Human | 23,391 | Homo sapiens
|
| L. infantum | 7,924 | Leishmania infantum
|
| M. jannaschii | 1,773 | Methanocaldococcus jannaschii
|
| Mouse | 21,615 | Mus musculus
|
| M. tuberculosis | 3,988 | Mycobacterium tuberculosis
|
| Asian rice | 43,649 | Oryza sativa
|
| P. falciparum | 5,187 | Plasmodium falciparum
|
| Rat | 21,272 | Rattus norvegicus
|
| Budding yeast | 6,040 | Saccharomyces cerevisiae
|
| pombe Fission yeast | 5,128 | Schizosaccharomyces
|
| S. aureus | 2,888 | Staphylococcus aureus
|
| T. cruzi | 19,036 | Trypanosoma cruzi
|
| Maize | 39,299 | Zea mays
|
Predicts single proteins, not complexes
- The AlphaFold 2 code predicts a structure for a single sequence.
- If single protein predictions are fit to an experimental assembly they often will have badly positioned
domains rigidly shifted by 5 to 50 Angstroms.
- Packing of proteins in an assembly effects the positions of domains that single protein predictions get wrong.
- Wrong domain positions can be corrected by splitting the alphafold models into domains and fitting each domain into a cryoEM map.
- AlphaFold-Multimer can predict small complexes, just released week ago.
AlphaFold-Multimer
AlphaFold-Multimer bioRxiv article 2021 code was released November 2, 2021.
- Predicts structures of assemblies by concatenating sequences of the assembly proteins and running single-sequence AlphaFold prediction, treating the assembly as one big fusion protein.
- Problem is that the multiple alignment of experimental sequences does not include sequences spanning multiple proteins.
- Contacts between residues are inferred from residue covariation seen in multiple sequence alignment, so without sequences that span multiple proteins there is no protein-protein contact information.
- AlphaFold-Multimer tries to remedy this by concatenating experimental sequences in the alignment.
Uses cross-chain pairing method method described in this
bioRxiv article from 2018
that uses separate methods for prokaryotes and eukaryotes.
- Dimer interface prediction success rate is about 65%.
Predicts hetero-multimers and homo-multimers, about 2/3 of pairwise interfaces correct
in a test on 4400 recent PDB complexes (2018-2021, less than 9 chains, less than 1500
total residues, clustering when all chains have > 40% identity, 2600 unique chain pair interfaces).
- Currently uses monomer structure templates but not multimer structures in making predictions.
Example AlphaFold-Multimer Predictions
Incorrect prediction (right, Figure 5 from AlphaFold-Multimer article) shows that the
AlphaFold predicted alignment error (confidence estimate for residue-residue distances) reveals
low-confidence in relative placement of two proteins.
Four examples of correct AlphaFold-Multimer predictions.

Figure 4 (from AlphaFold-Multimer bioRxiv article) | Structure examples predicted with the AlphaFold-Multimer. Visualised are the ground truth structures (blue) and predicted structures (coloured by chain).
|
Example of wrong AlphaFold-Multimer prediction.
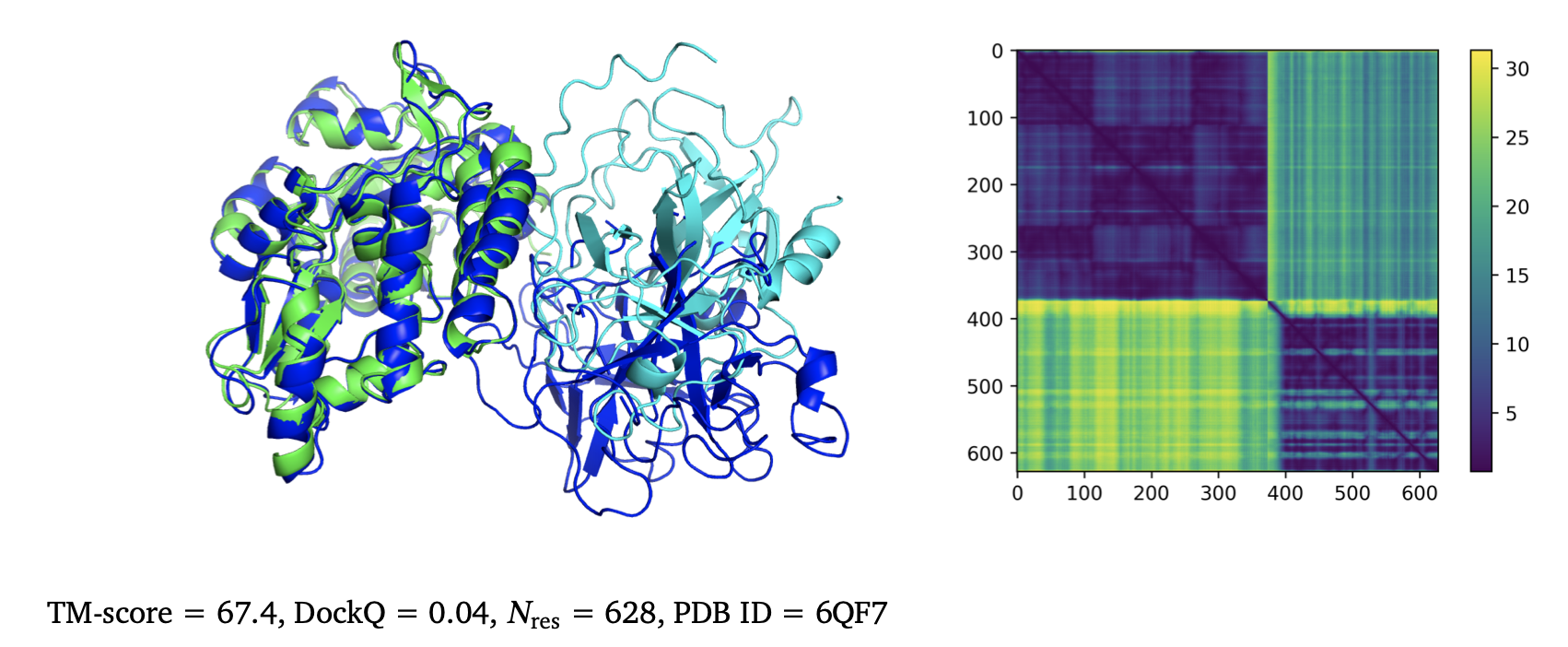
Figure 5 (from AlphaFold-Multimer bioRxiv article) | Example of a predicted heterodimer with incorrect geometry that is correctly predicted as low confidence by the predicted aligned error (PAE). Visualised are the ground truth structures (blue), predicted structures (coloured by chain), and PAE heat map. The PAE heat map shows the predicted error (in Angstroms) between all pairs of residues. |
How AlphaFold-Multimer concatenates experimental sequences for covariation analysis
- Cross-chain sequence associations are chosen by taking sequences from same species.
- For eurkaryotes most similar sequence to target for each chain are associated, then second
most similar sequence for each chain are associated, ....
- For prokaryotes uses a heuristic that associates sequences for each chain that have
lexicographically similar uniprot accession ids. Apparently this correlates with
proximity of genes on the full genome and for prokaryotes interacting proteins are
often close to each other in the genome (expressed as a group in operons). Looking
at alphafold code (data/msa_pairing.py) it really does just turn the uniprot id
into a single number (converting letters A-Z to digits) and compare difference.
The premiss is that the ids are assigned in order and depositors deposit proteins
in the order the appear in the genome -- seems an absurd heuristic.
- Cross-chain pairing method for eukaryotes may be not so good according original authors
of that method. Eukaryotes are harder due to paralogs and long genome separations.
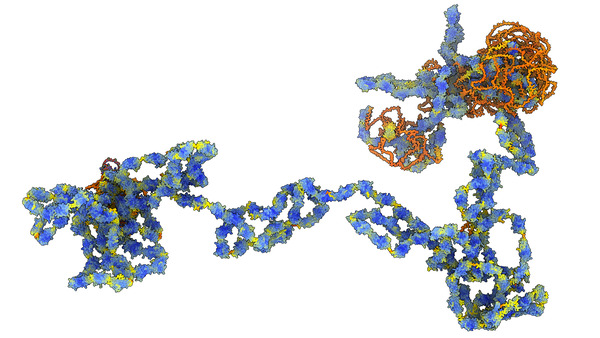
Human muscle protein Titin, 34000 amino acids,
pieced together
from 29 segment AlphaFold models.
|
Runs out of GPU memory for long sequences
- AlphaFold fails for longer sequences 800 - 2500 amino acids depending on amount of available GPU memory (typically 16 - 80 Gbytes).
- Maximum of ~800 amino acids for ChimeraX AlphaFold predictions on Google Colab using old GPUs (Nvidia K80, T4) with 16 Gbytes memory.
- Sequence length limitation is particularly restrictive for AlphaFold-Multimer where it applies to the sum of lengths of all sequences.
- When it fails it does not say "out of memory" -- the error messages are generally not useful but all the cases I have seen seem to be caused by out of memory. I have seen it fail on sequences as short as 100 amino acids when there were a very large number of aligned sequences (200,000) so it is not only the sequence length that controls memory use.
- The EBI AlphaFold database handle sequences up to length 2800 amino acids. They were computed by
using the combined memory of 4 GPUs.
- Longer human sequences were computed in 1400 amino acid chunks spaced every 200 amino acids.
- Long sequences also take long compute times, 20 hours for a 1600 amino acid myomesin-1 sequence
(UniProt MYOM1_HUMAN)
on an Nvidia A40 GPU with 48 Gbytes of memory.
AlphaFold does not handle ligands, ions, solvent
- If a protein conformation depends on whether a ligand (e.g. ATP) is bound, AlphaFold does allow
specifying ligands.
- AlphaFold will not predict the unbound form. If the PDB database has many examples of the bound
form the AlphaFold training may predict the bound form but without the ligand.
- It may be possible to predict liganded and unliganded conformations by giving AlphaFold liganded or unliganded structure templates.
- ChimeraX AlphaFold prediction capability does not use structure templates.
- The standard AlphaFold execution script does not allow specifying which templates to use -- only a directory of PDB models to search for templates. Specifying specific templates would require writing some custom Python scripts to start AlphaFold.
Requires expensive Nvidia GPU to run
- AlphaFold requires a modern high-end Nvidia GPU (uses CUDA) and Linux.
This isn't strictly true. This
AlphaFold issue says it can
be run in CPU-only mode with run_docker.py flag --use_gpu=false, but this may run 10 times slower.
- The GPU should have lots of memory, minimum 8 Gbytes, but 16 - 80 Gbytes allows handling longer sequences.
- The higher memory GPUs are specifically for non-graphics use and costly:
| Approximate Nvidia GPU prices
|
|---|
| Model | Memory | Price
|
|---|
| A100 | 80 Gbytes | $17000
|
| A100 | 40 Gb | $12000
|
| A40 | 48 Gb | $7000
|
| RTX 3090 | 24 Gb | $2000
|
| RTX 3080 | 12 Gb | $1000
|
- ChimeraX uses Google Colab which is free but use is limited (~2 hours per day) and GPUs have small memory, typically nvidia K80 or T4 with 16 Gbytes.
- Other cloud GPU services start at about $1 per hour for a lower end GPU with 16 Gb, to $3/hour for an A100 with 40 Gb (Google cloud).
Uses large databases
- AlphaFold uses large databases, 2 Tbytes, that can take days to download.
- For fastest AlphaFold computations the databases might be best on fast disk (e.g. NVMe drives).
- Keeping databases up to date may also be an issue for longterm use of AlphaFold.
- ChimeraX AlphaFold on Google Colab uses reduced databases (15 times smaller) and streams them to the cloud machine for each computation.
- Using paid cloud services with full AlphaFold databases requireslarge persistent disk cloud storage, or reduced databases streamed for each computation requires network bandwidth which may add substantial costs, setup and maintenance.