Tom Goddard
October 10, 2023
Predict protein-protein interactions by running AlphaFold dimer predictions of all pairs of sequences and seeing which ones are predicted with high confidence.
The David Baker lab demonstrated this approach search for complexes in yeast identifying 106 previously unidentified assemblies from 5495 dimer predictions:
Computed structures of core eukaryotic protein complexes.
Humphreys IR, + 29 more authors
Science. 2021 Dec 10;374(6573)
Hiten Madhani gave me 31 protein sequences involved in gene silencing and pH-sensitive signaling in fungus Cryptococcus neoformans. Many are suspected to interact with one another but there are few experimental structures.
"The RNAi, DNA methylation, and Polycomb components files all relate to gene silencing. I anticipate interactions within these sets (indeed, by IP-MS we know of several complexes and interactions) and possibly between them.
The RIM101 set is a pH-sensitive signaling pathway distantly related to the vertebrate Hedgehog signaling pathway. Almost nothing is known about protein-protein interactions in this system.
Finally, in the Polycomb and DNA methylation files, I have included the sequences of histone H3 and H4. I am curious whether any of the components interact with the H3-H4 tetramer complex as a histone chaperone at the replication fork as this is currently a hot and interesting area. For them, I suggest a 3-chain prediction: Factor X: H3: H4."
There are 31 genes listed below, sequence length in parentheses. Note CLR3 appeared in both DNA methylation and polycomb sets.
| RNAi components (10) | AGO1 (927), GWO1 (1641), GWC1 (584), QIP1 (747), RDE1 (1476), RDE2 (1238), RDE3 (426), RDE4 (481), RDE5 (396), RDP1 (1123) |
| DNA methylation components (8) | CLR2 (714), CLR3 (731), DMT5 (2377), MIT1 (952), MIT2 (1160), SUV420 (1120), SWI6 (222), UHRF1 (342) |
| Polycomb components (6) | BND1 (1595), CCC1 (976), CLR3 (731), EED1 (570), EZH2 (729), MSL1 (435) |
| Histones H3, H4 | HHT1 (138), HHF1 (103) |
| RIM101 pathway (6) | RIM101 (916), RIM13 (813), RIM20 (902), RIM23 (488), RRA1 (654), RRA2 (743) |
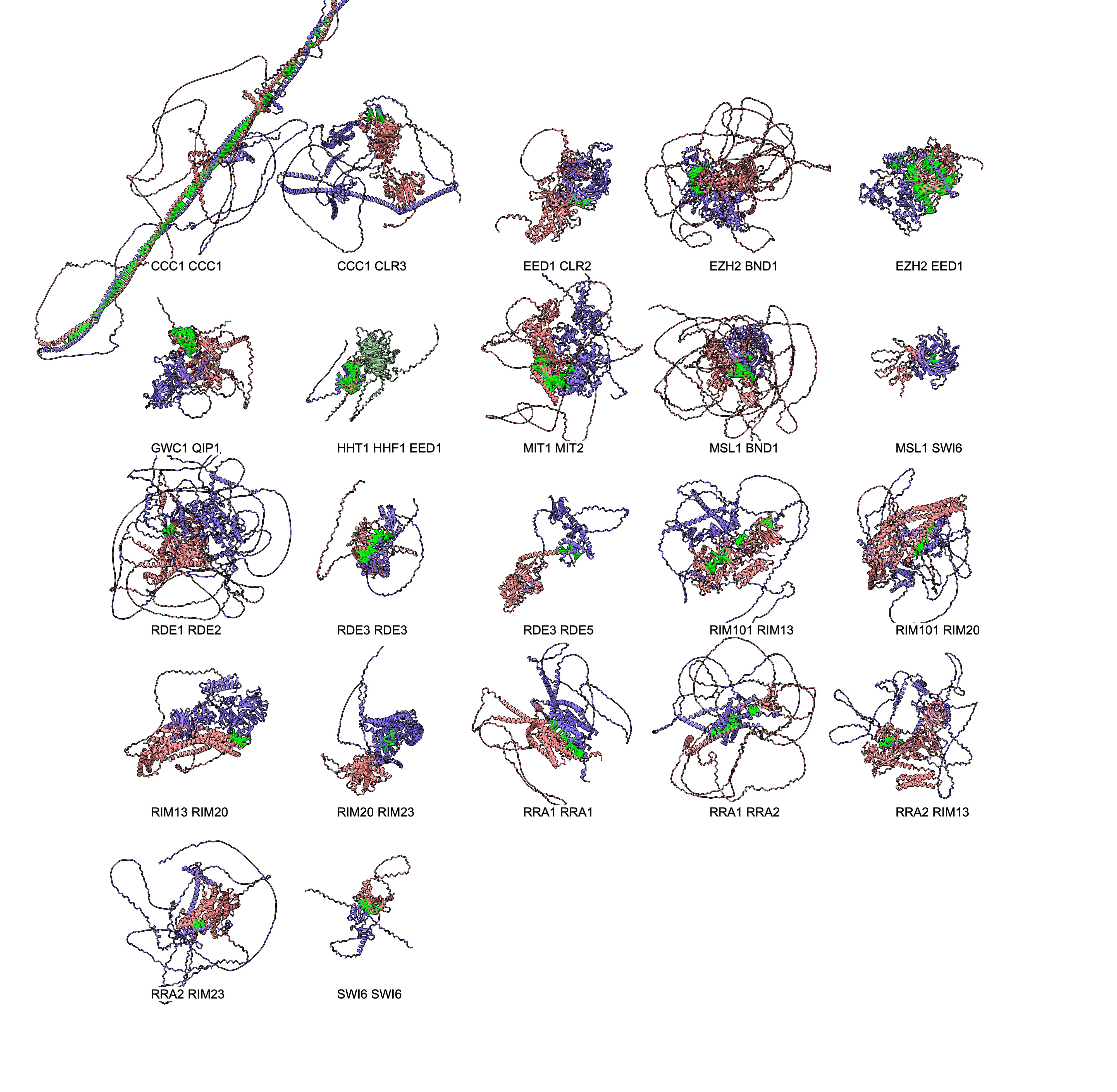
Made AlphaFold predictions for all 31 monomers, took 9 hours. These are colored by the standard AlphaFold predicted local distance difference test with blue being high confidence and yellow, orange, red low confidence. This image was made with the ChimeraX tile command and the labels.py script.

Aim was to run 310 Alphafold predictions:
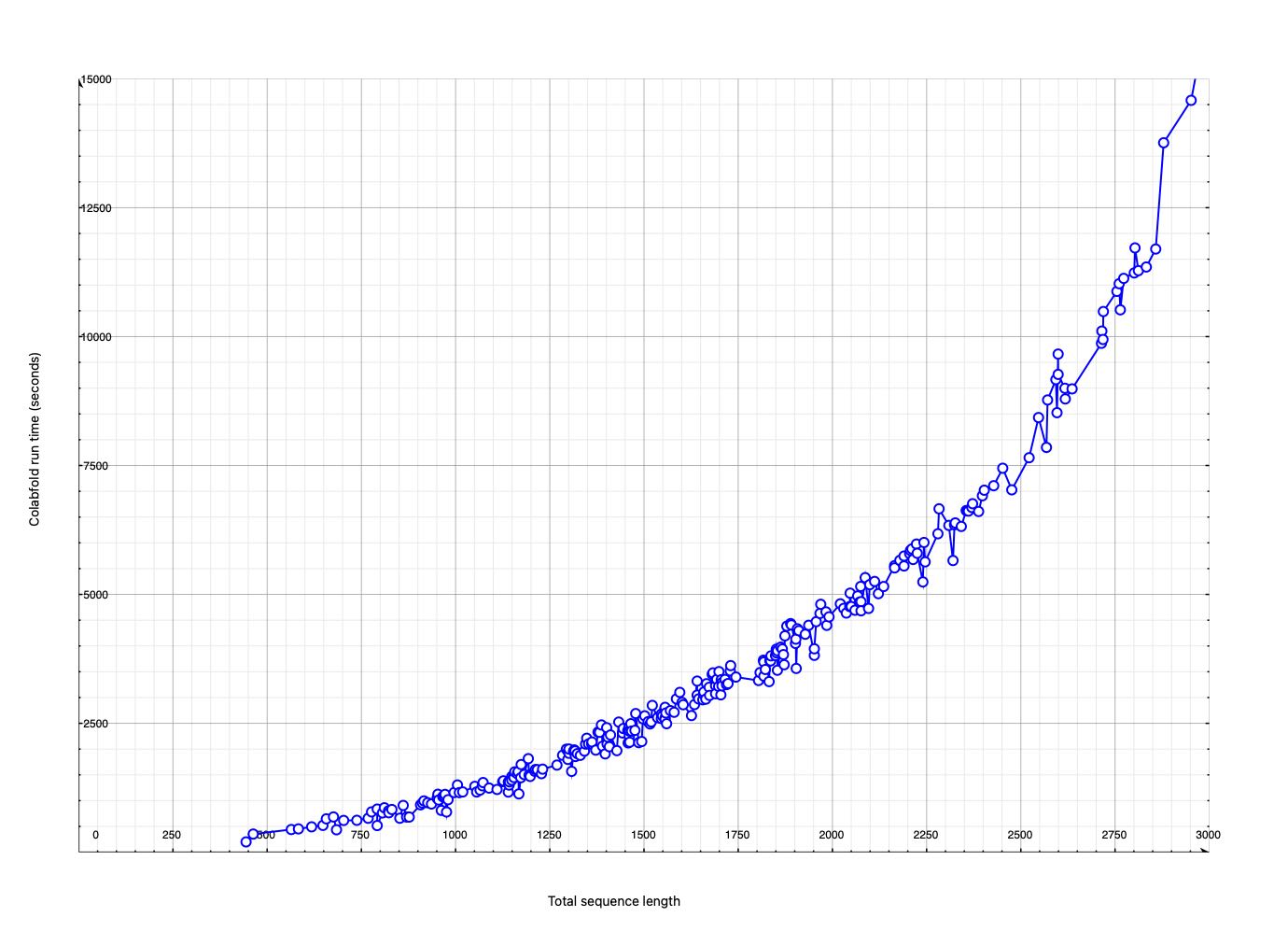
Completed 290 (93%) Alphafold predictions in 10 days on a single Nvidia 3090 GPU.
Did not run 20 that had total sequence length > 2900. Tried GW01 RDE1 dimer with 3117 residues and it crashed, apparently due to the 24 GByte memory of the Nvidia 3090 GPU being too small.
I've started running the 6 remaining non DMT5 dimers, October 30, 1 pm. The last ~2900 length jobs took only 4 hours, so should take a bit more than a day to finish these 6. Only the two smallest completed apparently due to limited GPU memory (24 GB on Nvidia 3090).
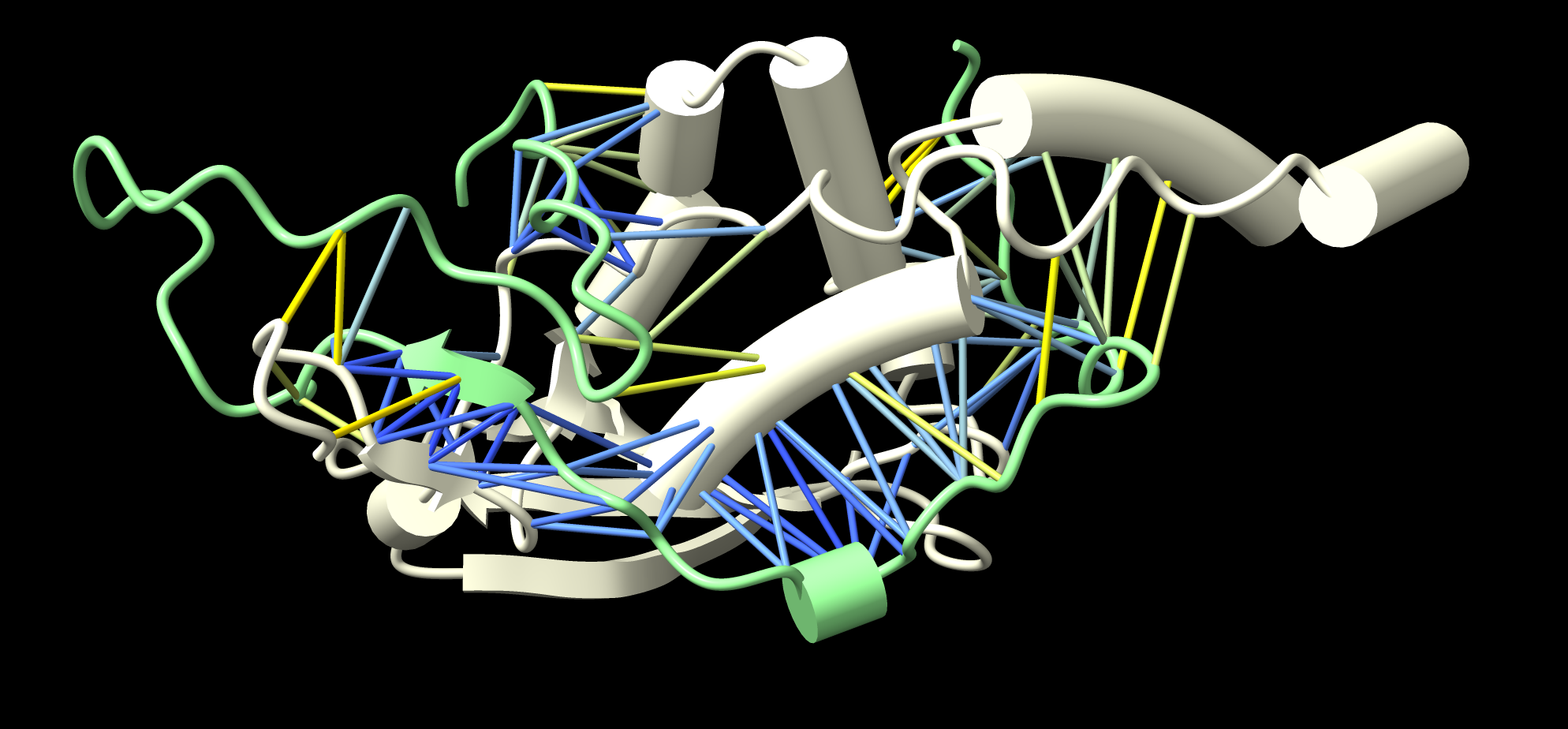
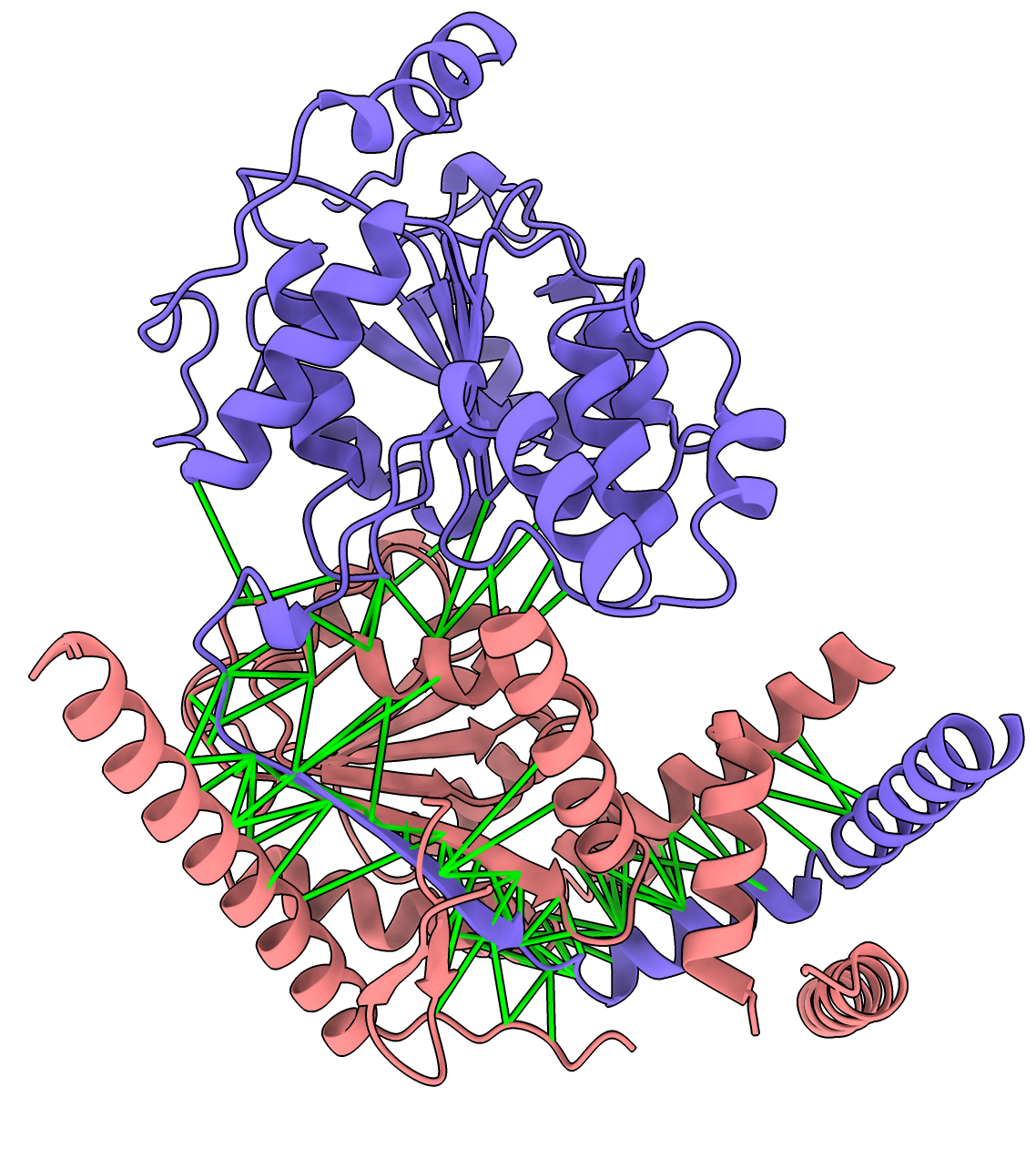
Here are 22 predicted interactions from the 290 AlphaFold predictions. These are ones where the AlphaFold predicted aligned error between contacting (< 4A) residues had high confidence values (< 5A) for 10% or more of the close interface residue-residue pairs. List of results dimers_best.csv. Dimers are pink and blue with green cylinders between low PAE interface residue pairs. All the interfaces are surprisingly small. The image was made with the ChimeraX tile command and interface.cxc script to show the green lines for high confidence PAE interactions.
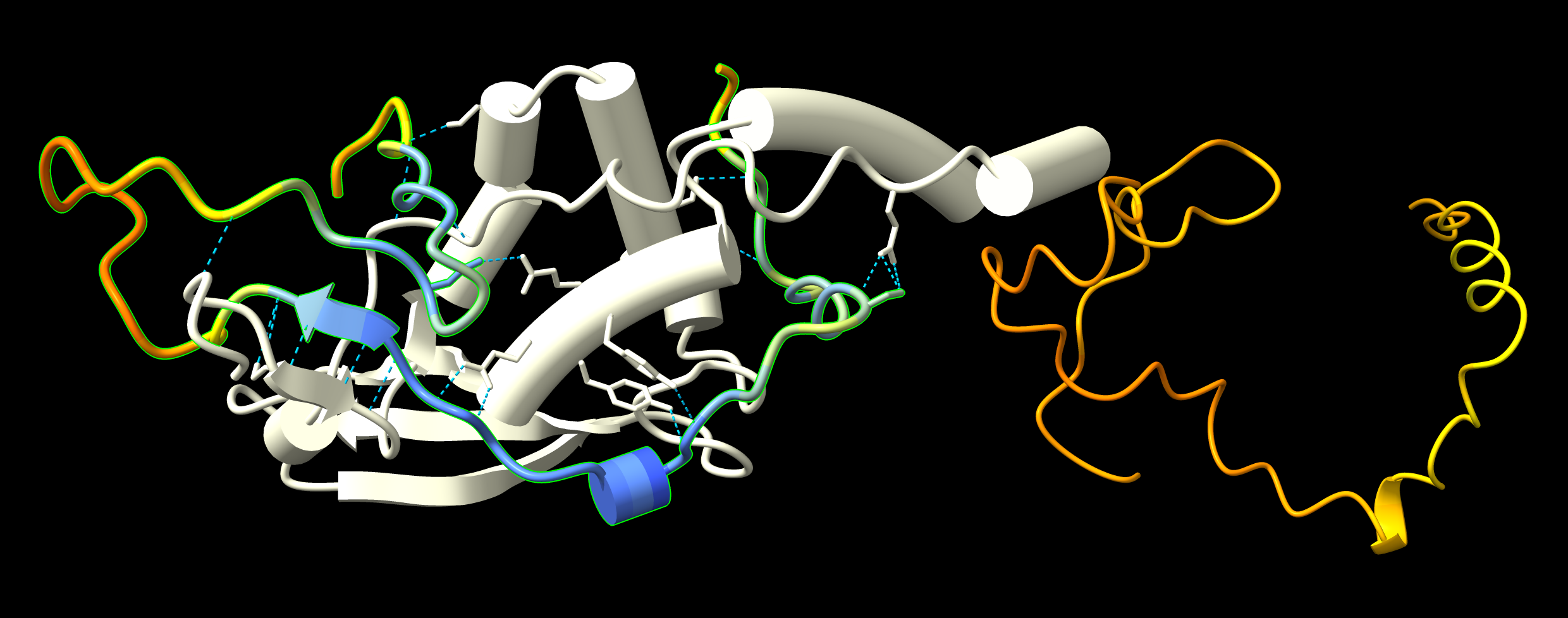
Here is more detail on one of the predicted dimers GWC1 and QIP1 with only the inteface residues shown.
|
|
|
| GWC1 to QIP1 binding PAE values less than 5 Angstroms. | Predicted hydrogen bonds GWC1 to QIP1. Monomer GWC1 prediction at right has low pLDDT confidence (orange), while bound form has high confidence (blue) regions. |
I searched the PDB for structures having homologs of pairs of the proteins. I found 1506 PDB entries involving 3554 PDB chains with homology to the 31 sequences using a BLAST E-value cutoff of 1e-3. For each PDB I looked if two of the 31 search sequences are found (possibly two copies of the same sequence) and in those cases I check if the two PDB chains have residues in contact within 4 Angstroms. This produced 2400 different PDB dimers. Imposing a BLAST cutoff of 1e-9 and minimum number of 10 interface residues (the smaller of the residues in the two chains within 4 Angstroms of the other chain), then there are 20 pairs of the 31 sequences that have PDB homologs shown in this graph from Cytoscape. The PDB with lowest E-value match is shown in the graph.
The homologs in the PDB may differ from our 31 Cryptococcus neoformans proteins.
All of these interactions may be wrong and require verification.
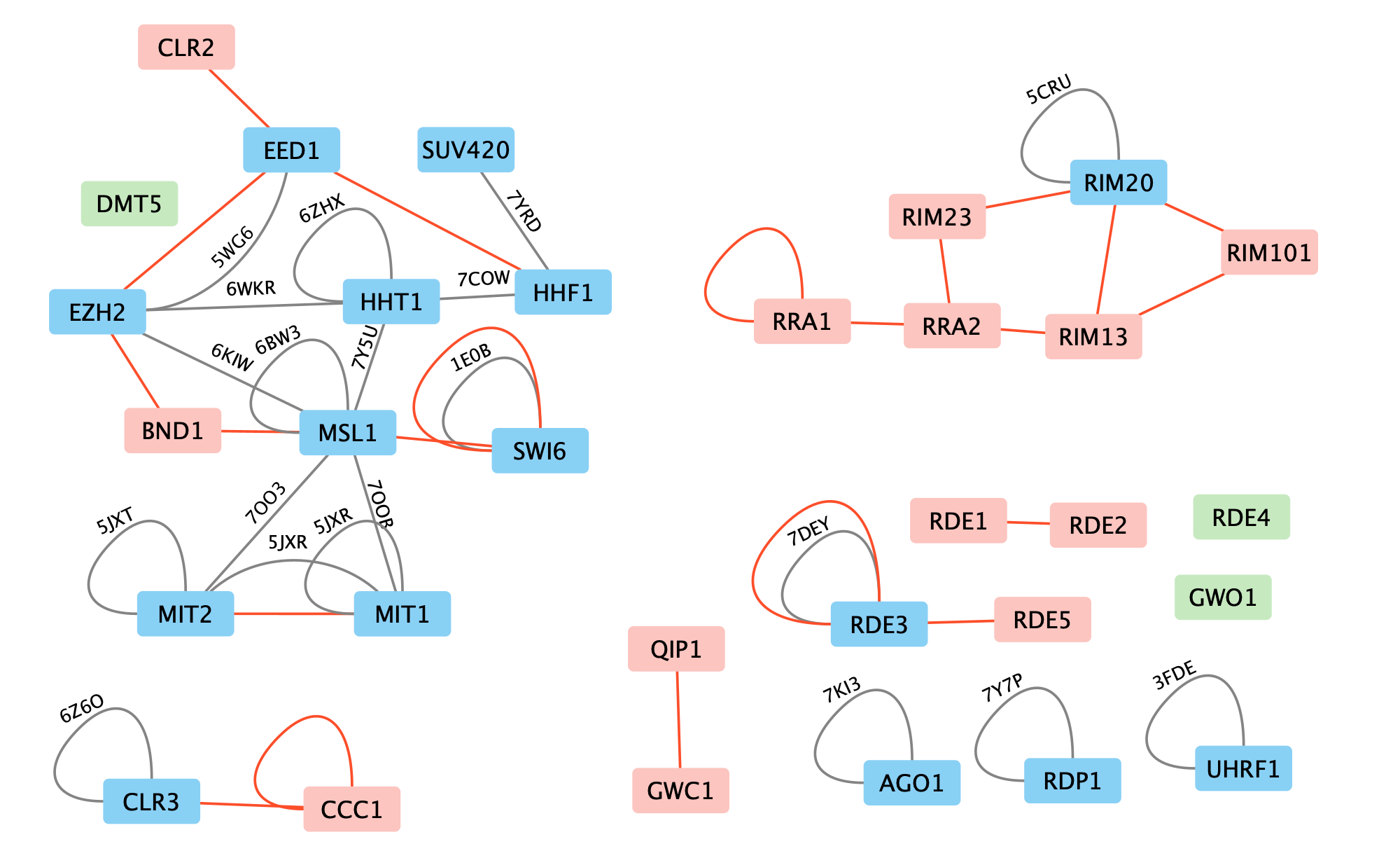
4 of the 22 Alphafold predictions (blue-pink) have PDB experimental structures (gray). Three of the four agree.
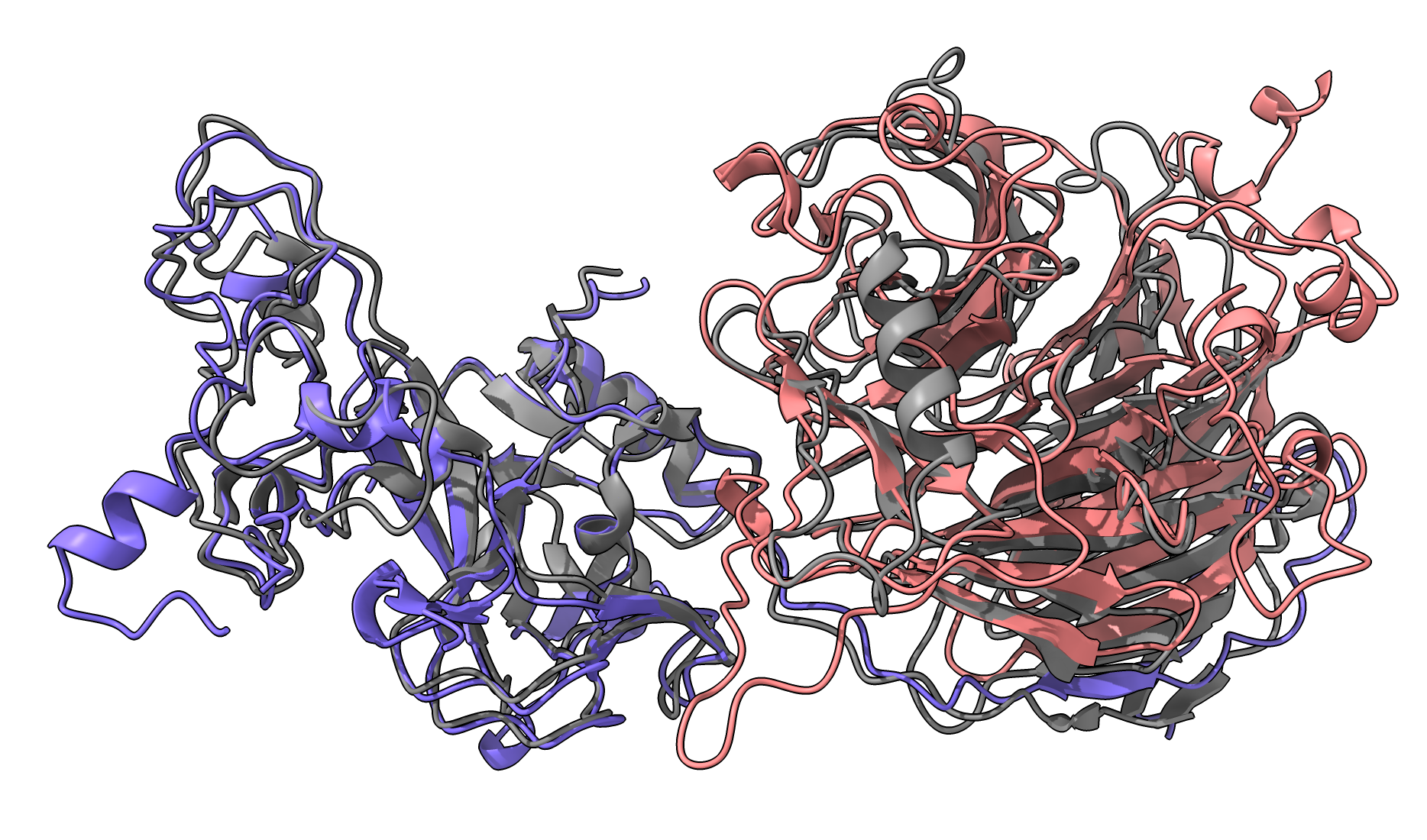 EZH2-EED1 purple-pink, 8fyh gray Other PDB entries 4W2R 5BJS 5HYN 5IJ7 5IJ8 5KJH 5KJI 5KKL 5LS6 5M5G 5TQR 5VK3 5WF7 5WFC 5WFD 5WG6 6B3W 6C23 6C24 6KIU 6KIV 6KIW 6KIX 6KIZ 6PWV 6PWW 6W5I 6W5M 6W5N 6WKR 7KSO 7KSR 7KTP 7MBM 7MBN 7TD5 7UD5 8FYH |
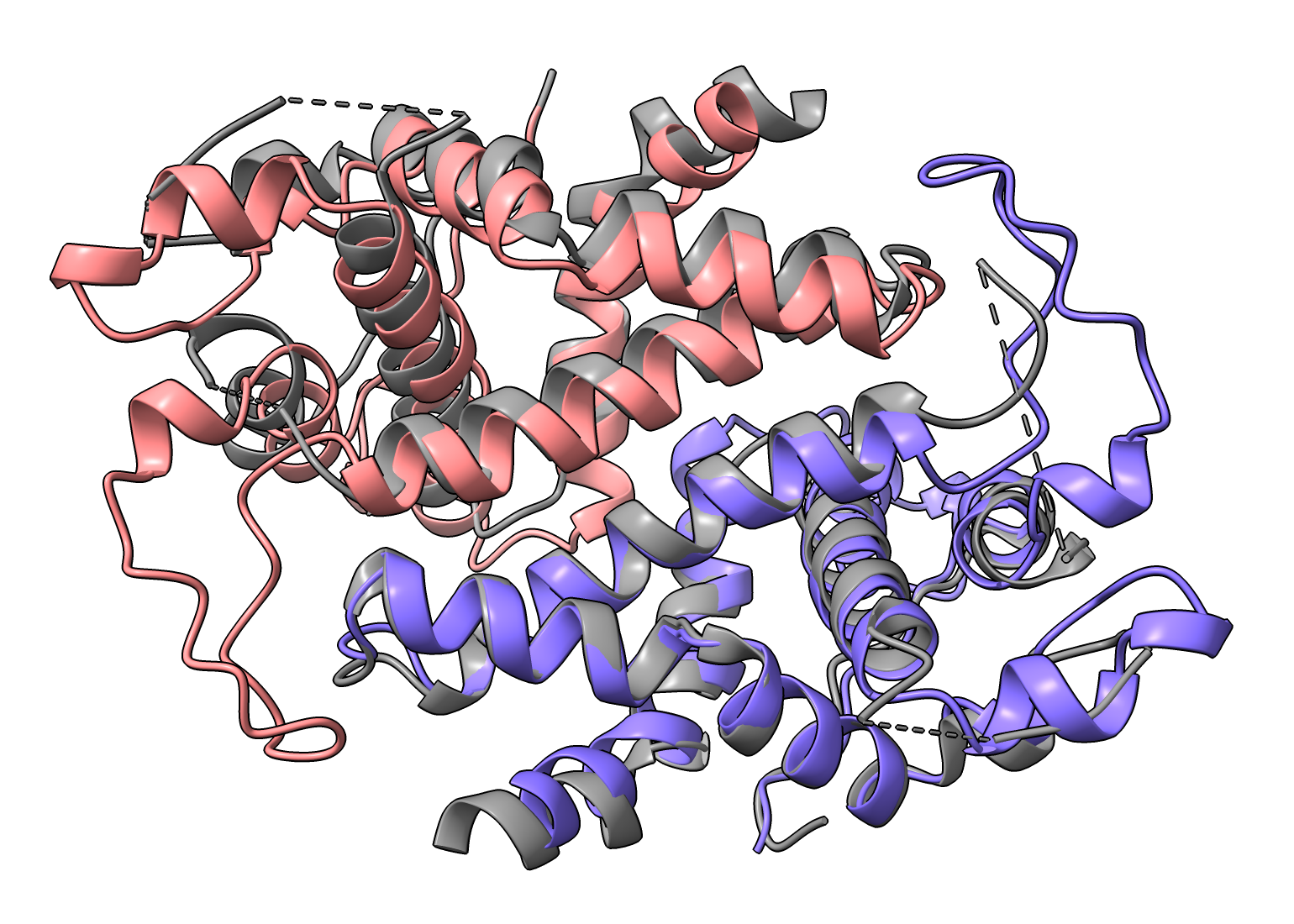
RDE3-RDE3 purple-pink, 7dey gray Other PDB entries 1O0W 2FFL 2QVW 3RV0 3RV1 5T16 6V5B 7DEY 7R97 |
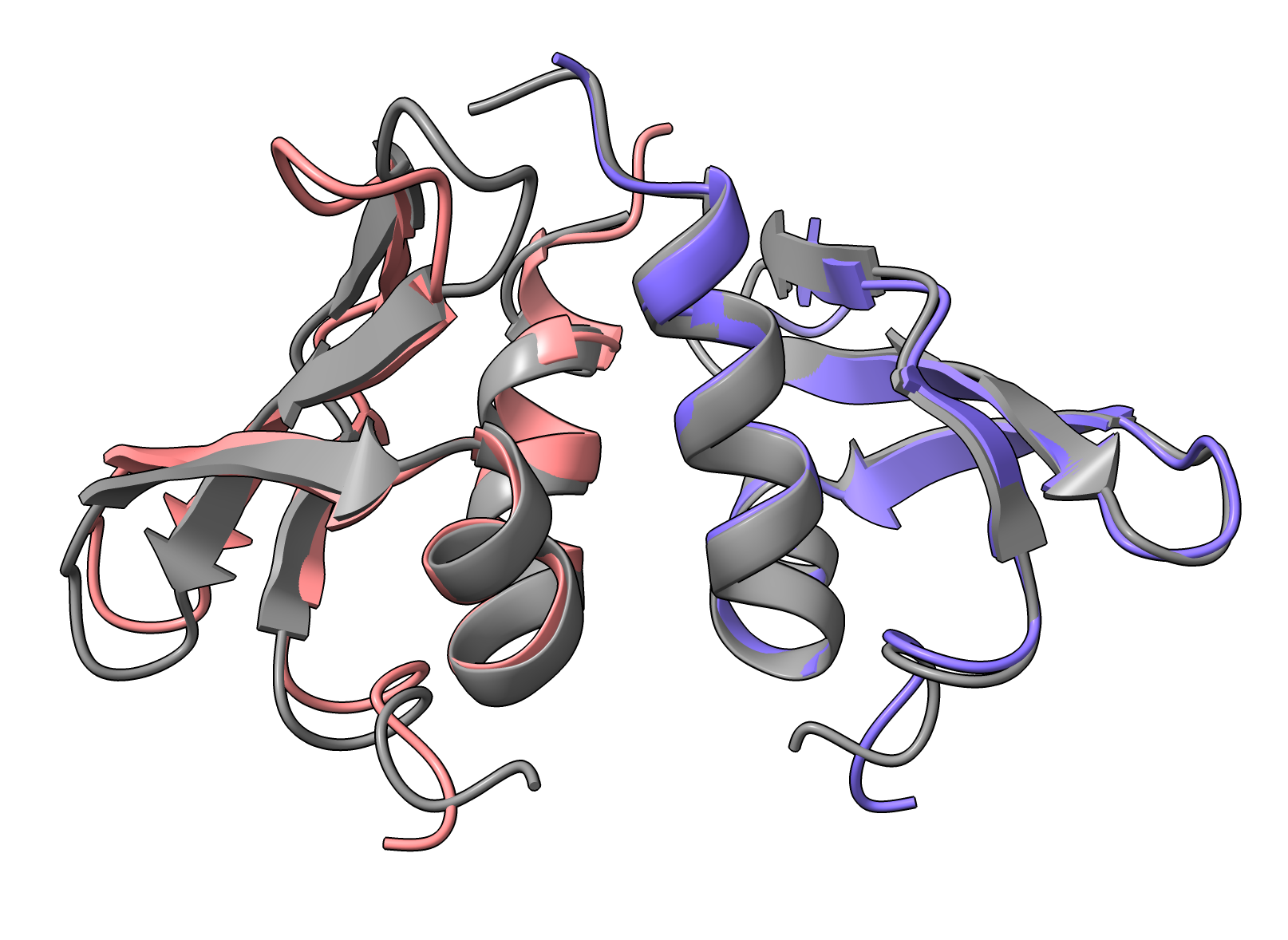 SWI6-SWI6 purple-pink, 1e0b gray
SWI6-SWI6 purple-pink, 1e0b grayOther PDB entriess 1E0B 3DM1 3H91 3I90 3I91 3LWE 3QO2 3R93 4FSX 4FT2 4FT4 4QUF 4U68 4X3K 4X3S 4X3T 4X3U 5EPL 6D07 6D08 6FTO 6V2D 6V2H 6V2S 6V3N 6V8W 7N27 7SLW 7VRF) |
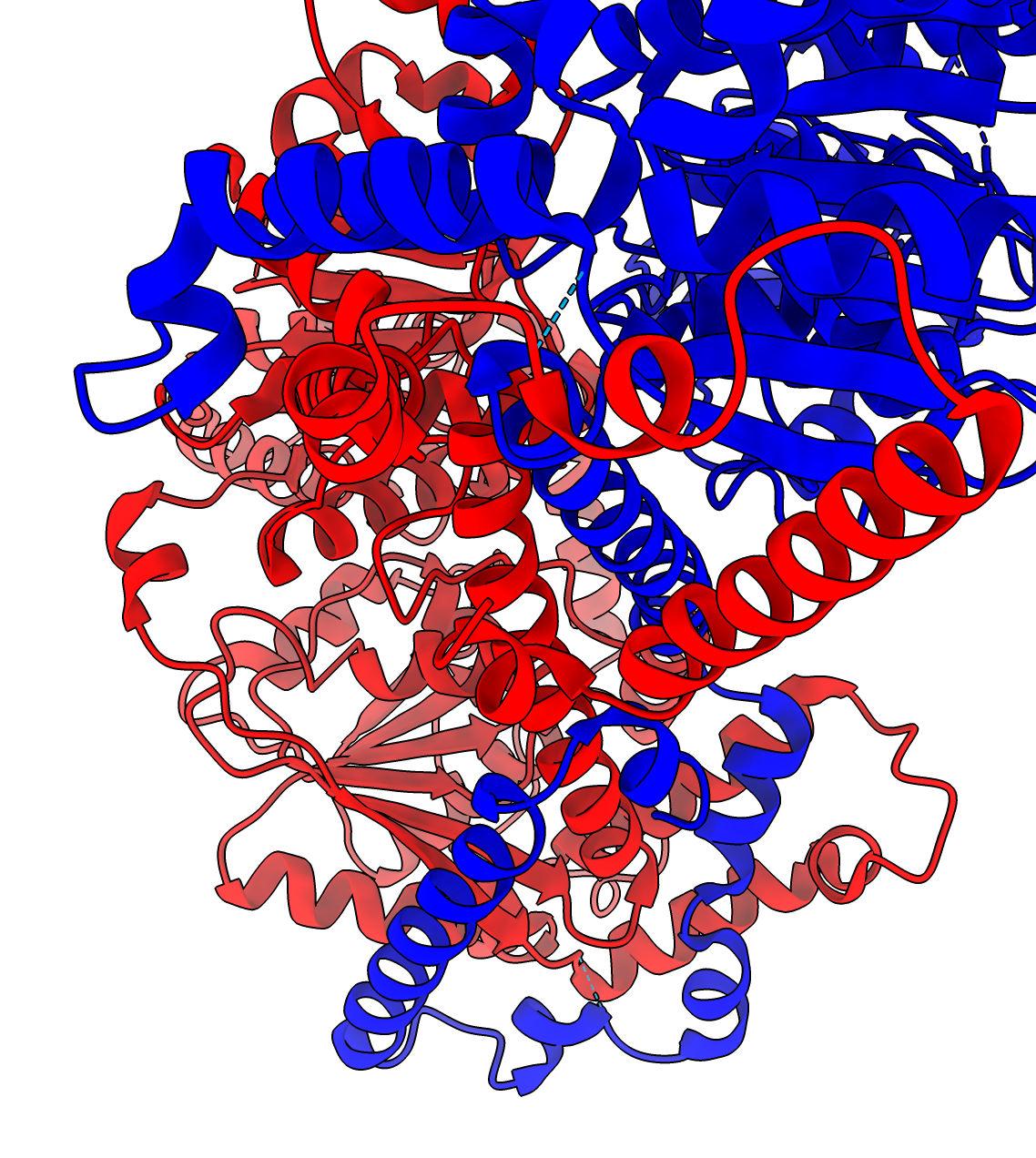
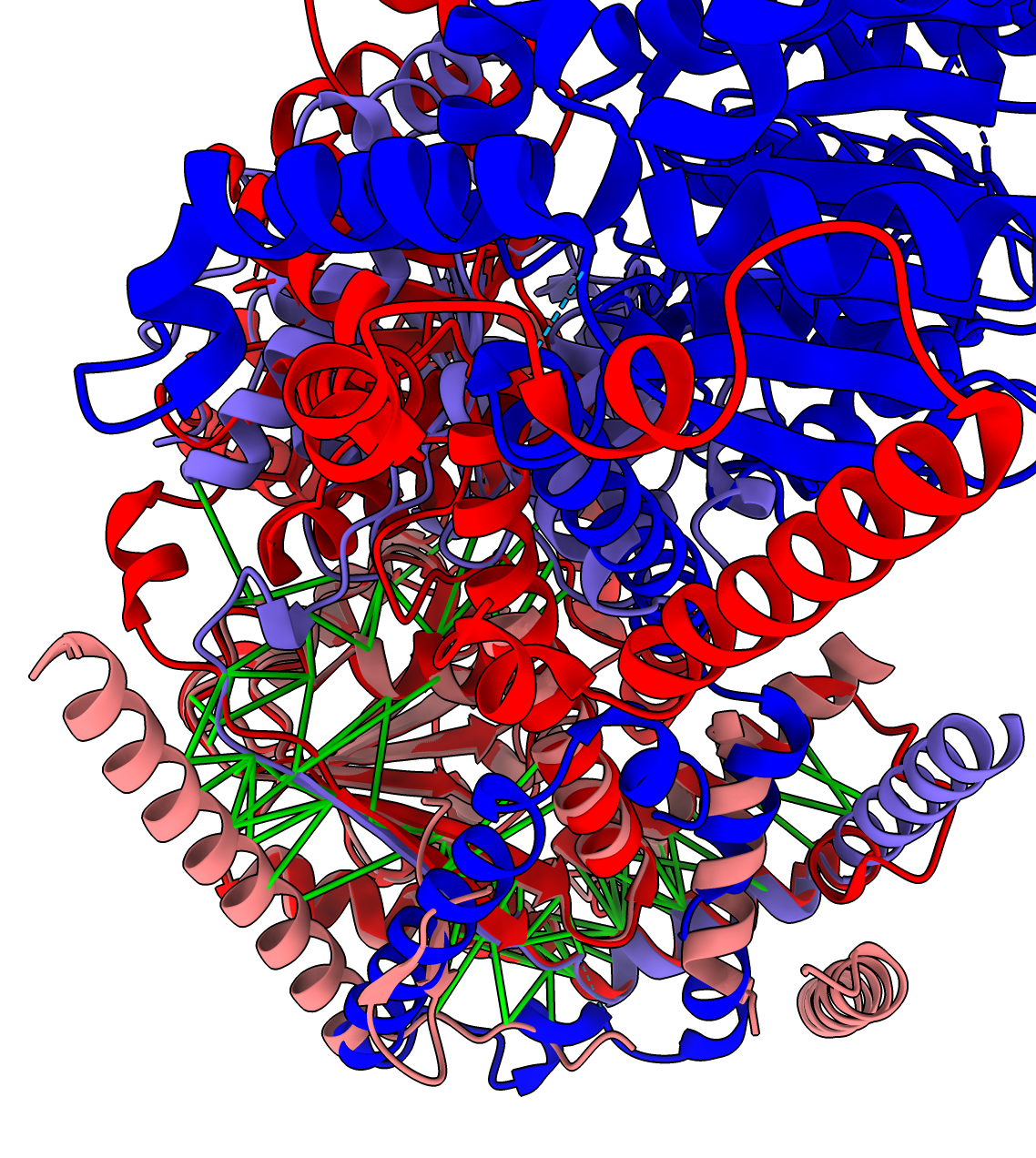
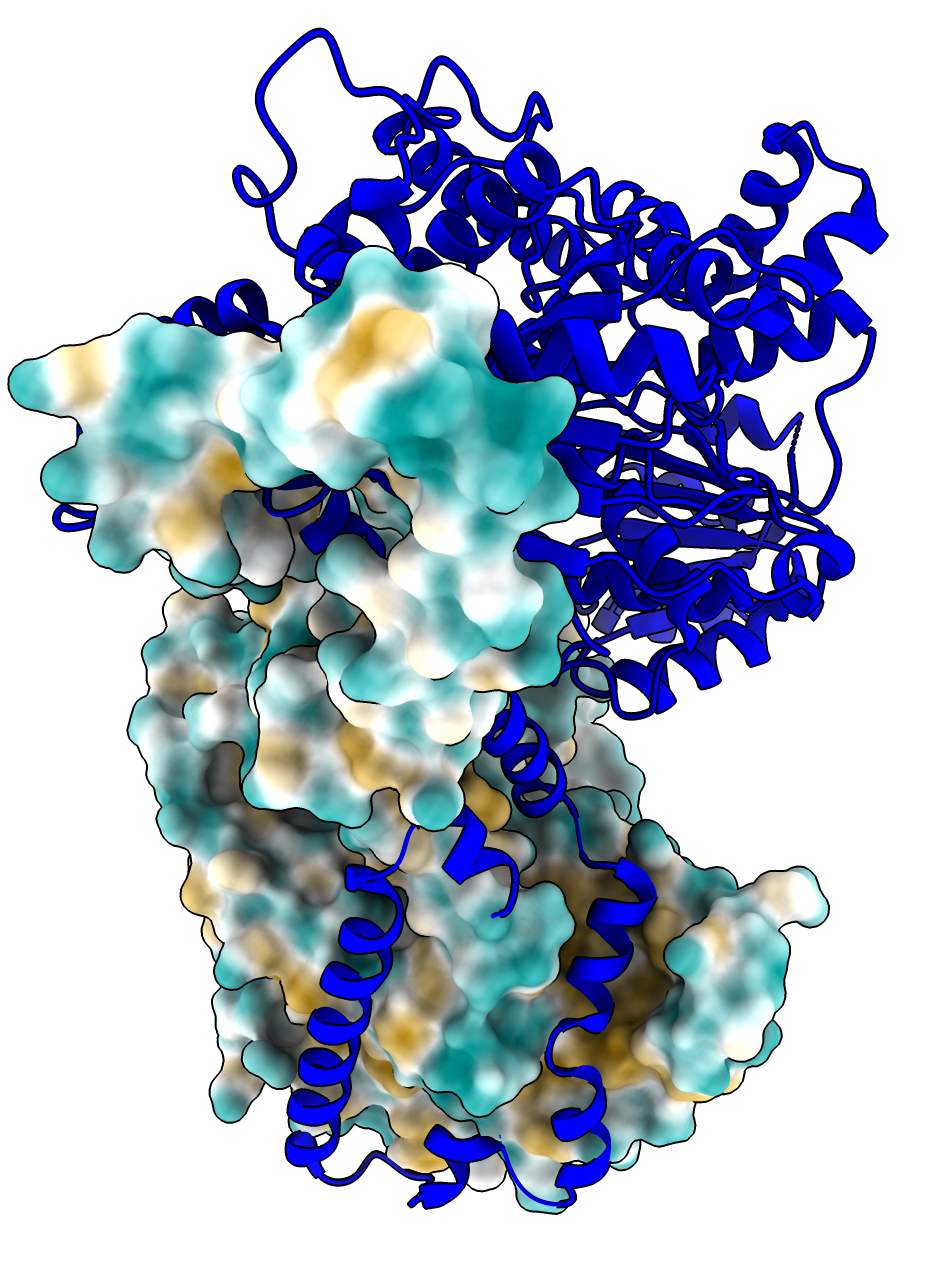
For 1 of the 4 AlphaFold predictions where a PDB experimental dimer exists, they don't match. The MIT1 MIT2 AlphaFold asymmetric dimer is different from the PDB 5jxr experimental chromatin remodeling homodimer complex. The dimerization residues seen in the experimental homodimer exist in MIT1 but not in MIT2, so this PDB experimental model is not a good template for MIT1 and MIT2 dimerization.
Details: The MIT1 and MIT2 sequences have lengths 952 and 1160 but only residues 656-928 of MIT1 and 15-230 of MIT2 align with the PDB 5jxr model with BLAST Evalues 6e-48 and 6e-41. Only the MIT1 sequence has the dimerization subsequence of the 5jxr homodimer. This shows that our PDB dimer search needs to take account of whether the sequence alignment between our target and the PDB entry includes the PDB dimerization region.
| 
| 
| 
|
| MIT1 MIT2 AlphaFold dimer interface with high confidence PAE in green. | PDB 5jxr dimer. | Superposed Alphafold and PDB models shows the AlphaFold predicted interface is all in one monomer in the PDB model (Alphafold purple helix and strand matches red PDB helix and strand). | PDB 5jxr hydrophobic surface on one monomer shows helix of one monomer sits in hydrophobic groove of other monomer. |
I tried running the 21 RIM101 dimers using 20 AlphaFold recycles instead of the default 3 since this has been reported to give better multimer predictions. There were 8 RIM101 dimer interactions found with 3 recycles and there were 8 found with 20 recycles. Both calculations found dimers: RIM101.RIM13, RIM101.RIM20, RIM13.RIM20, RIM20.RIM23, RRA1.RRA1, RRA1.RRA2, RRA2.RIM23, while only the 3 recycles found RRA2.RIM13 and only the 20 recycles found RRA1.RIM13. I wrote a ChimeraX script dimers_align.py to align the 7 dimers that both predicted using the common interface residues and all 7 had low RMSD C-alpha of 0.08 to 1.36 Angstroms. The 20 recycles calculation took 69 hours on the Nvidia 3090 GPU while the 3 recycles took 14 hours, so about 5 times slower. This makes sense since 3 recycles means 4 passes, while 20 recycles amounts to 21 passes, so about a factor of 5 more passes. A few of the predictions did not use all 20 recycles because apparently it stops based on a convergence criteria at fewer cycles and the specified number is the maximum.
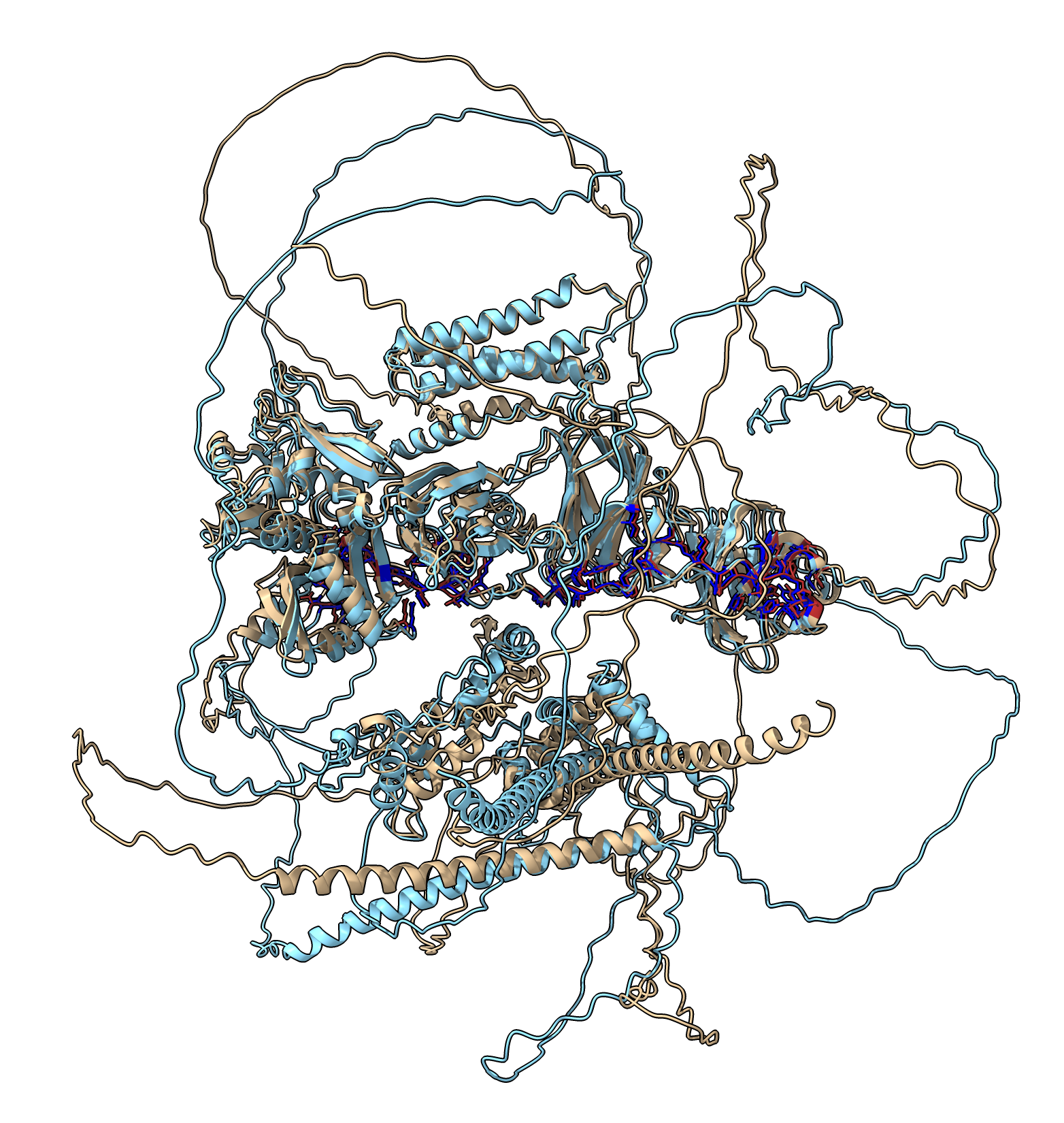
| Alignment of 20 recycle (tan) and 3 recycle (light blue) RIM101 RIM13 dimers. Interface residues in dark blue and red. RMSD 1.36 Angstroms. |
Could the predictions be done much faster without much loss of accuracy using 1 recycle and predicting just one model instead of 5. The following test shows we only got half the 3-recycle interfaces.
I ran the 21 RIM101 dimer predictions with these settings and it took 1.5 hours. It found 5 dimers RIM101.RIM13, RIM13.RIM20, RIM20.RIM23, RRA1.RRA1, RRA1.RRA2. All 5 of these were found with the 3 recycle and 20 recycle runs. Three align with the 3 recycle results with low RMSD values, one has a high RMSD of 5 Angstroms, and one did not align at all because the interface residues were all different. Here are the RMSD values RIM101.RIM13 5.09, RIM13.RIM20 no match, RIM20.RIM23 0.327, RRA1.RRA1 1.13, RRA1.RRA2 0.5. The 5 Angstrom RMSD is caused by their being two interfaces in separate domains that can move relative to each other. Aligning each domain gives RMSDs 0.46 and 1.28 Angstroms.
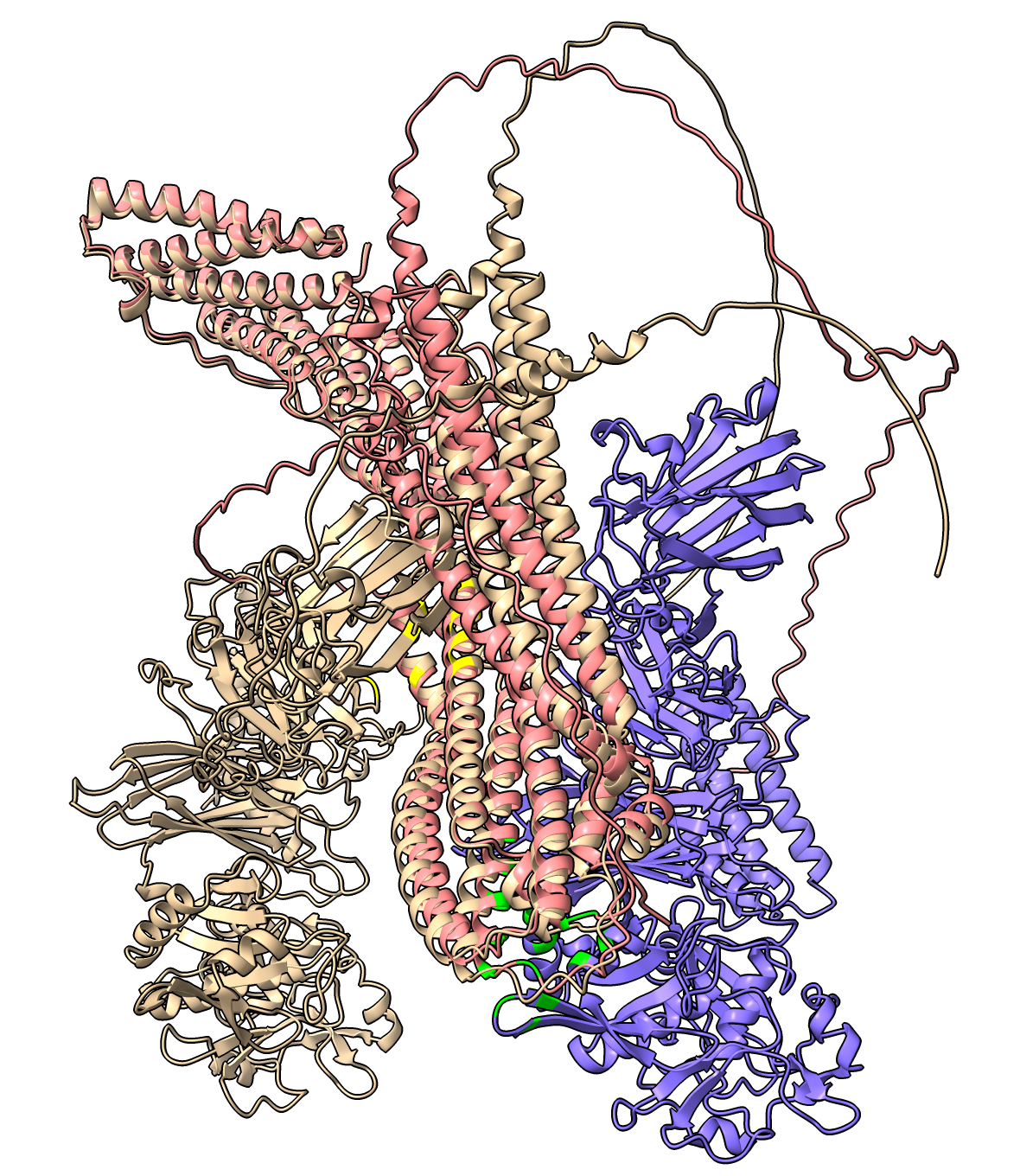
| Alignment of 1 recycle (tan) to 3 recycle (blue/pink) RIM13 RIM20 predictions with interfaces (yellow and green) involving entirely different residues. |
I tried the smallest dimer RIM23 RIM23, total length 976, only using the CPU (by setting environment variable VISIBLE_CUDA_DEVICES=-1), 1 recycle, 1 model. It took 104 minutes on an Intel i9-13900K (3 GHz, 24 cores, minsky.cgl.ucsf.edu, 64 GB memory). On an older i9-9900KF (3.6 GHz, 8 cores, quillian.cgl.ucsf.edu, 64 GB) it took 126 minutes. It was using 5 cores the whole run (500% CPU reported by top). I did not specify the number of cores, perhaps this is the jax default. By comparision with the Nvidia 3090 GPU it on quillian it took 1 minute 39 seconds. So 5 CPU cores was 63 times slower than 3090 GPU, and 1 CPU core would be expected to be 315 times slower.
There are a lot of ideas of ways to do this better. The basic questions are:
Original sequence files from Hiten
Derived sequence files
AlphaFold predictions produced 5 models per prediction. In the zip files below I only include one to save space. And for the dimers I only include the 22 where the interface confidence is high.
To run all the AlphaFold predictions I used localcolabfold from Github following the Linux installation instructions on the Github web page on October 10, 2023. I installed on an Ubuntu 22.04.3 LTS system with Nvidia 3090 GPU driver 535.113.01 cuda 12.2, 24 GB graphics memory, Intel i9-9900KF CPU, 64 GB main memory, host quillian.cgl.ucsf.edu. The 3090 is rated at 350 watts, and nvidia-smi showed it using 200-340 watts during prediction.
Monomer predictions were run with this command
nohup colabfold_batch all_monomers.fasta . >& all_monomers.out &
Multimer predictions were run with
nohup colabfold_batch all_multimers.fasta --num-recycle 3 . >& all_multimers.out &
To run only model 2 of the 5 AlphaFold models with just 1 recycle
nohup colabfold_batch all_multimers.fasta --num-recycle 1 --model-order 2 . >& all_multimers.out &
The fasta file for multimers has the sequences for a prediction separated by colons.
Times for predicting 5 models with 3 recycles using colabfold_batch on Nvidia 3090.