last updated May 2026
This tutorial shows how to color a structure by the conservation in a multiple sequence alignment. In principle, any sequence alignment that ChimeraX can read and can associate with the structure of interest can be used for conservation coloring. However, the required number and diversity of sequences in the alignment depend on what you are trying to illustrate. See also: Color Key, Profile Grid conservation labels, online sources of sequence alignments
|
|
|
The three parts of this tutorial can be done independently.
Start ChimeraX. If you want to use the click-to-execute links in this page, view it in the ChimeraX Browser, e.g. by entering the ChimeraX command:
Command: open https://www.rbvi.ucsf.edu/chimerax/data/conservation-coloring/conservation-coloring.html
Fetch 121p, a structure of H-Ras, from the RCSB Protein DataBank:
Command: open 121p
Hide water and show the ligand as ball-and-stick:
Command: hide solvent
Command: style ligand ball
If you wish, review how to manipulate structures (part of the Binding Sites tutorial).
The sequence-alignment file can be fetched directly from our website with the following command, OR you can download 121p-consurf.aln as plain text to a convenient location on your computer and use the menu: File... Open to open it.
Command: open https://www.rbvi.ucsf.edu/chimerax/data/conservation-coloring/121p-consurf.aln

|
The Sequence Viewer can be a separate window, or it can be docked into the combined ChimeraX window (details...). The sequence window can be moved/docked/undocked by dragging its top bar, and resized by dragging its edges (if docked) or corners (if undocked). The image shows it docked into the same area as the Models panel so that the two are tabbed. This was done by dragging the sequence window and hovering it over that area. Clicking a tab brings the corresponding panel to the front.
The first sequence in the alignment automatically associates with the structure, as indicated by the colored box around its name: Input_pdb_SEQRES_A. The sequence has this name because the structure was submitted to the ConSurf Server to obtain the alignment.

|
A histogram above the sequences indicates the conservation per column. These conservation values are automatically assigned to the residues of any associated structures as an attribute named seq_conservation. The default method for calculating conservation is the entropy-based measure from AL2CO (a program included with ChimeraX courtesy of Pei and Grishin).
Try the default coloring, where the entire range of values in the structure is spread across the palette:
Command: color byattr seq_conservation
The coloring palette can be specified as a series of colon-separated color names or by any of several built-in palette names:
Command: color byattr seq_conservation palette blue:red:yellow
Command: color byattr seq_conservation palette cyanmaroon
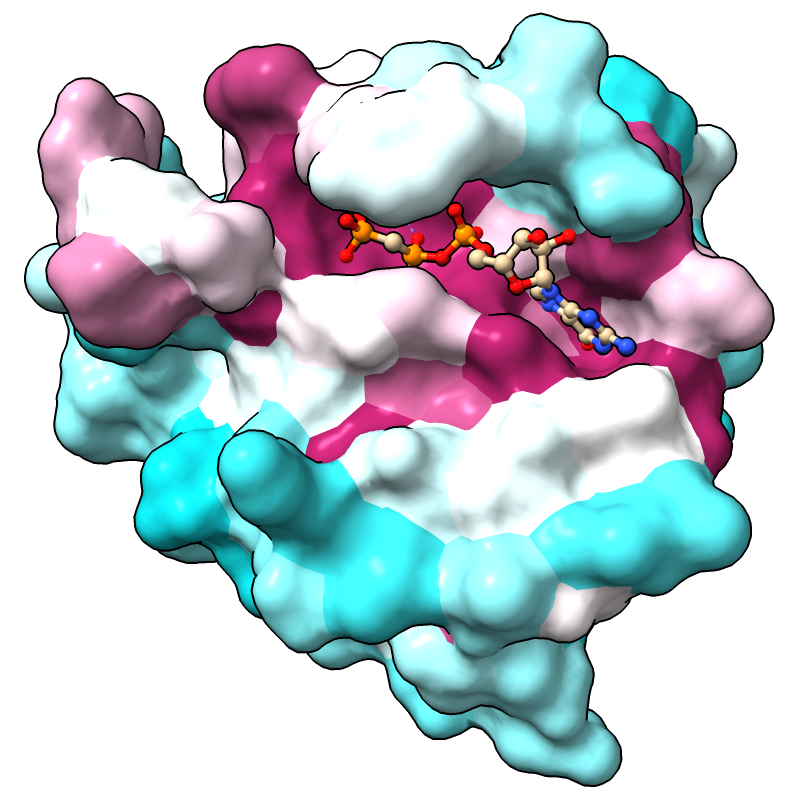
The full range of values is reported in the Log, in this case, -1.47 to 2.83 if the default method for calculating conservation is used. (Part 3 covers how to change the method.) For more emphasis on extreme values, the palette can be mapped to a narrower range, for example:
Command: color byattr seq_conservation palette cyanmaroon range -1.4,1.4
Positions with values below or above the specified range are given the first and last colors of the palette, respectively. The most highly conserved residues (maroon) are in the GTP-binding site.
Notice that cartoon segments at both the N- and C-termini of the protein have retained their original color, tan. Conservation values were not calculated for the corresponding positions in the sequence alignment because they have a high proportion of gaps. You can see this by scrolling the sequence window all the way to the left or right; you may also need to scroll up and down, as the alignment contains many sequences. (Part 3 covers how to change the gap tolerance.)

|
Any residues not associated with a sequence alignment, including solvent, ligands, ions, and other proteins, will also lack a conservation value. Residues without a conservation value can be assigned a color with the novalue option of the command, but to avoid recoloring ligands, etc., an atom specification may be needed to limit its scope of action. For example, in the following command, the coloring is limited to protein only:
Command: color byattr seq_conservation protein palette cyanmaroon range -1.4,1.4 novalue yellow
Attributes can also be used in command-line specification, for example, to show and label the residues with conservation values above some cutoff. By default, hide/show commands apply to atomic displays, not the cartoon:
Command: hide protein
Command: show ::seq_conservation>1.8
Command: label ::seq_conservation>1.8
Command: label height 1.3
Try the publication preset, with white background and black silhouette outlines:
Command: preset publication 1
For publication images, 2D labels (example in the last figure below) are nicer than these “3D” labels that move along with the structure. Remove the 3D labels:
Command: label delete
|
|
Molecular surfaces can also be colored with color byattribute, as above, or by simply applying the current atom coloring to the surface:
Command: surface
Command: color fromatoms
Use the interactive preset to return to a black background without outlines, and then close the structure and sequence alignment:
Command: preset inter
Command: close session
Fetch 3sn6, a structure of the β2-adrenergic receptor signaling complex:
Command: open 3sn6
The Log shows some information about the structure: chains A,B, and G are the trimeric G protein, chain N is a nanobody, and R is the receptor, actually a fusion with endolysin. The nonstandard residue is a high-affinity agonist of the receptor.
Use the cylinders preset to show cartoons with tube helices:
Command: preset cylinders
The receptor is a very dark green. Make it a brighter color and hide the nanobody:
Command: color /R yellow
Command: hide /N cartoons
The sequence-alignment file can be fetched directly from our website with the following command, OR you can download 81321.ali as plain text to a convenient location on your computer and use the menu: File... Open to open it.
Command: open https://www.rbvi.ucsf.edu/chimerax/data/conservation-coloring/81321.ali

|
The image shows the sequence window docked across the top of the overall ChimeraX window, in the same area as the Toolbar.
None of the sequences associated automatically with the receptor structure, as you can see by the absence of a colored box around any sequence name.
Command: sequence associate /R 81321.ali
Red boxes in the alignment (scroll horizontally to find them) indicate a couple of mismatches between the sequence in the structure and that in the alignment, but such small differences do not cause any problems for association.
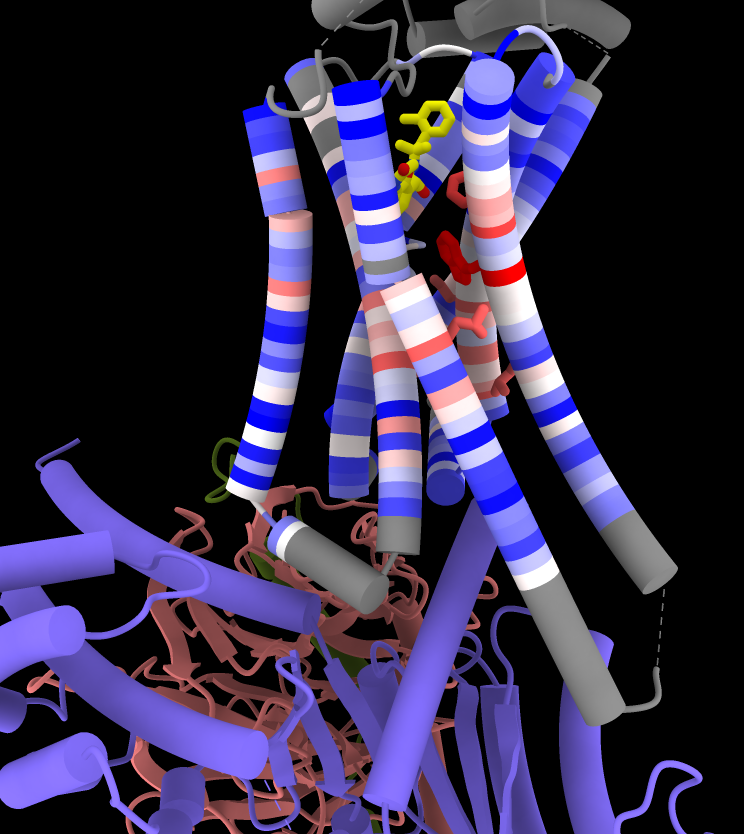
Now that the receptor is associated with the alignment, it can be colored by sequence conservation:
Command: color byattr seq_conservation /R & protein range -1.5,3 novalue gray
Command: show ::seq_conservation>2
Make sticks fatter and color the ligand by heteroatom:
Command: size stickrad .5
Command: color ligand byhet
The stark shadows in full lighting are somewhat distracting. Try soft lighting instead, which uses shadows from 64 directions to approximate ambient occlusion:
Command: light softThe tubes look a bit “smudgy,” especially in the single-color chains. Sometimes this smudginess can be reduced by using a greater number of directions for ambient shadows:
Command: light soft multishadow 512
The tradeoff for this improved appearance of ambient shadows, however, is that using a high number of directions may slow down the response to interactive manipulation. Return to simple lighting, then close the structure and sequence alignment:
Command: light simple
Command: close session
Fetch 1kmo, an outer-membrane protein of E. coli, and hide atoms so that only the cartoon is shown:
Command: open 1kmo
Command: hide
The sequence-alignment file can be fetched directly from our website with the following command, OR you can download 56935.ali as plain text to a convenient location on your computer and use the menu: File... Open to open it.
Command: open https://www.rbvi.ucsf.edu/chimerax/data/conservation-coloring/56935.ali

|
The image shows the sequence window docked across the top of the overall ChimeraX window, in the same area as the Toolbar.
The structure automatically associated with sequence d1kmoa-, as indicated by the colored box around its name in the sequence window. Color the structure by the values in the Conservation histogram above the alignment:
Command: color byattr seq_conservation
Much of the structure is still the original color (tan), including the interior β-sheet and many loops in the outer β-barrel. Conservation values were not calculated for these residues because they are in columns of the sequence alignment with a high proportion of gaps. The Conservation histogram is blank at these positions. Select residues that lack a conservation value:
Command: select ~::seq_conservationWe can change the conservation settings to allow more gaps, keeping in mind that there should still be enough residues in a column to provide a reasonably accurate measure of conservation.
Show the Sequence Viewer context menu (right-click or Ctrl-click on the sequence window, depending on platform) and choose Settings. The settings dialog can be left as a separate window or docked within the overall window, as described above for the Sequence Viewer itself. In the settings, switch to the Headers tab, which includes the conservation parameters. Changes in settings are applied immediately, so that the histogram bars will change right away. However, coloring by conservation must be reapplied to reflect the new values.
Increase the allowed Gap fraction to 0.6, then reevaluate which parts are still missing values:
Command: select ~::seq_conservation
Now much more of the structure is “covered” by conservation values. Clear the selection and reapply the coloring:
Command: select clear
Command: color byattr seq_conservation
The default method for calculating conservation is the entropy-based measure from AL2CO (a program included with ChimeraX courtesy of Pei and Grishin). AL2CO contains other measures (variance-based, sum of pairs) and options besides the Gap fraction. If you like, change any of the AL2CO parameters, see the histogram adjust, and reapply the coloring.
Click Save if you want to save the current settings as preferences. Click Reset to change the settings back to the “factory” defaults.
|
|
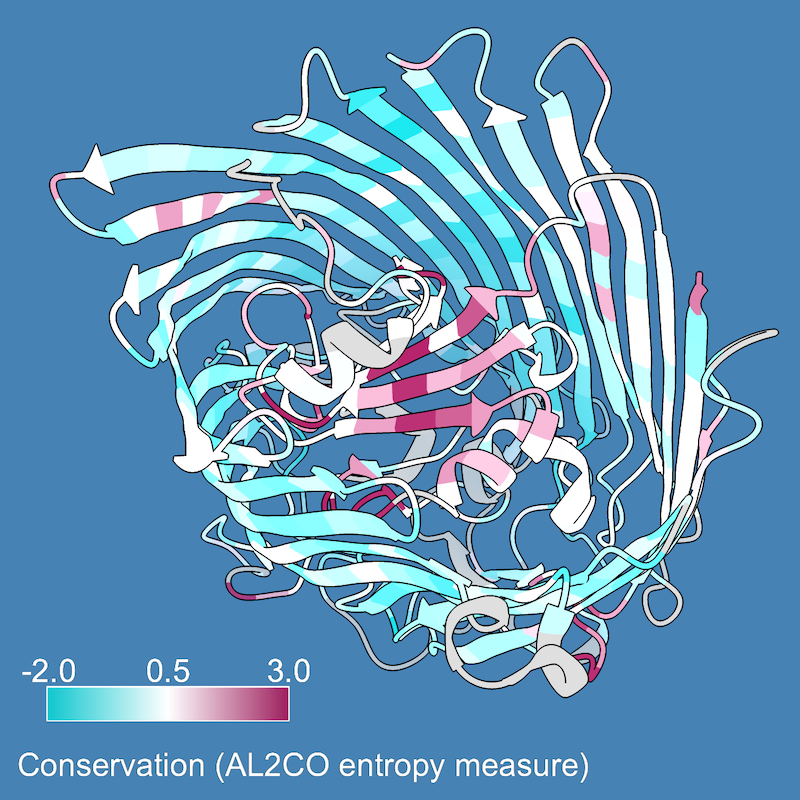
As in part 1, you may want to use different colors and/or apply them over a narrower range of values. Also, in ChimeraX 1.2 (May 2021) or newer, a color key can be added with the key option:
Command: color byattr seq_conservation palette cyanmaroon range -2,3 novalue silver key true
This draws an initial color key, but also opens the Color Key tool and enables redrawing and moving the key with the mouse (the Adjust key... option in the tool). For the example image, the key was redrawn on the lower left, and the Color Key Labels settings were used to put the numerical labels above instead of below the key. The Adjust key... option can be unchecked to restore the mouse to moving the structure instead of the key.
Other setup for the example image:
Command: windowsize 600 600
Command: preset publication 1
Command: set bg steelblue
Command: light flat
Command: graphics sil depth 0.03
A 2D label was added to explain the key:
Command: 2dlab text "Conservation (AL2CO entropy measure)"
...and the move labels
mouse mode
![]() .
was used to drag it to the desired location.
.
was used to drag it to the desired location.
Although it is usually most convenient to adjust the key settings and the key and label positions interactively as described above, commands could be used instead:
Command: key labelSide left/top
Command: key pos 0.06,0.1 size 0.3,0.04
Command: 2dlabels xpos 0.021 ypos 0.021
What sizes and positions to use for these annotations will depend on the dimensions of the graphics window.