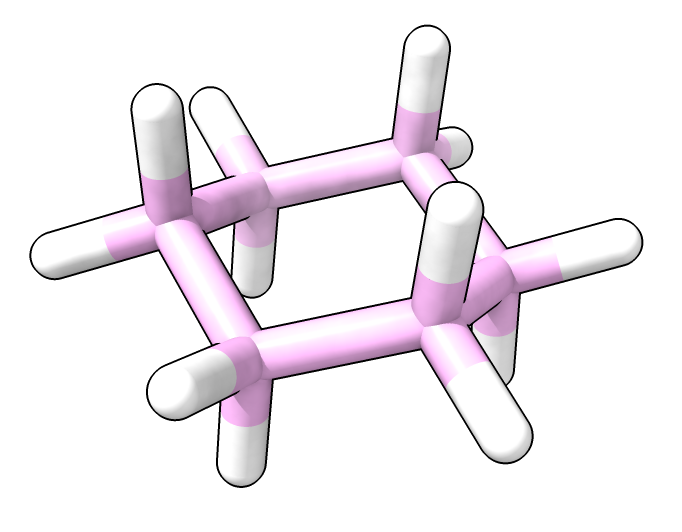
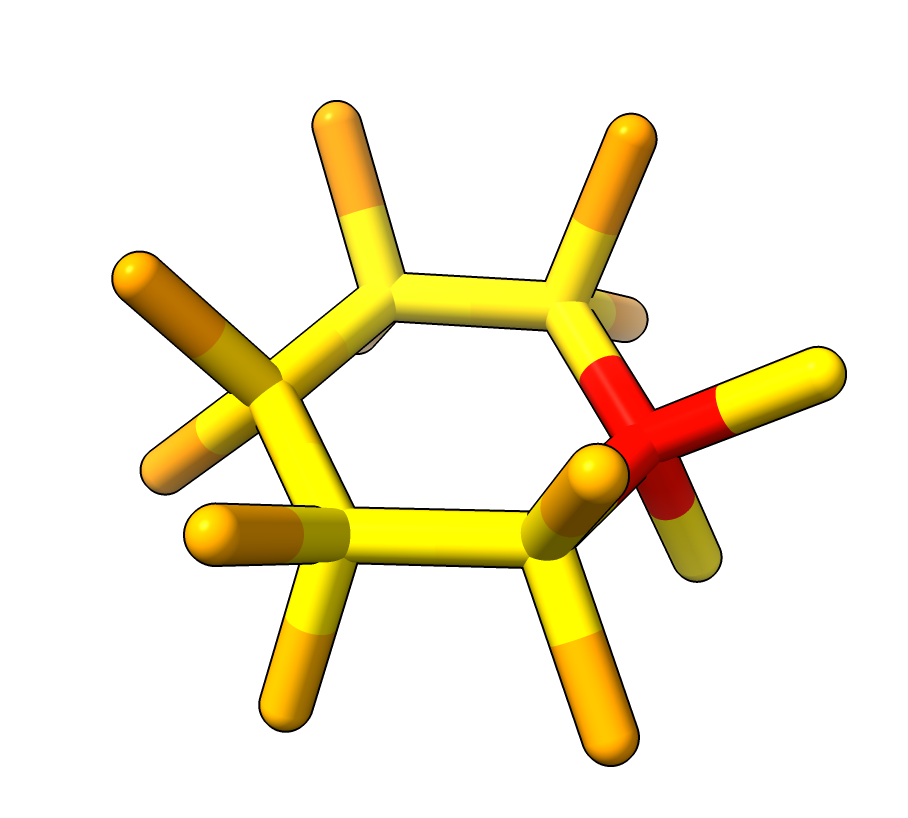
Cyclohexane (PubChem 8078).
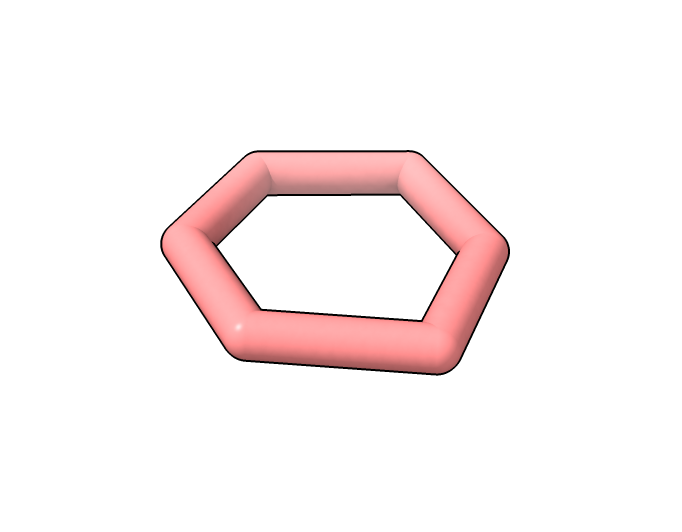
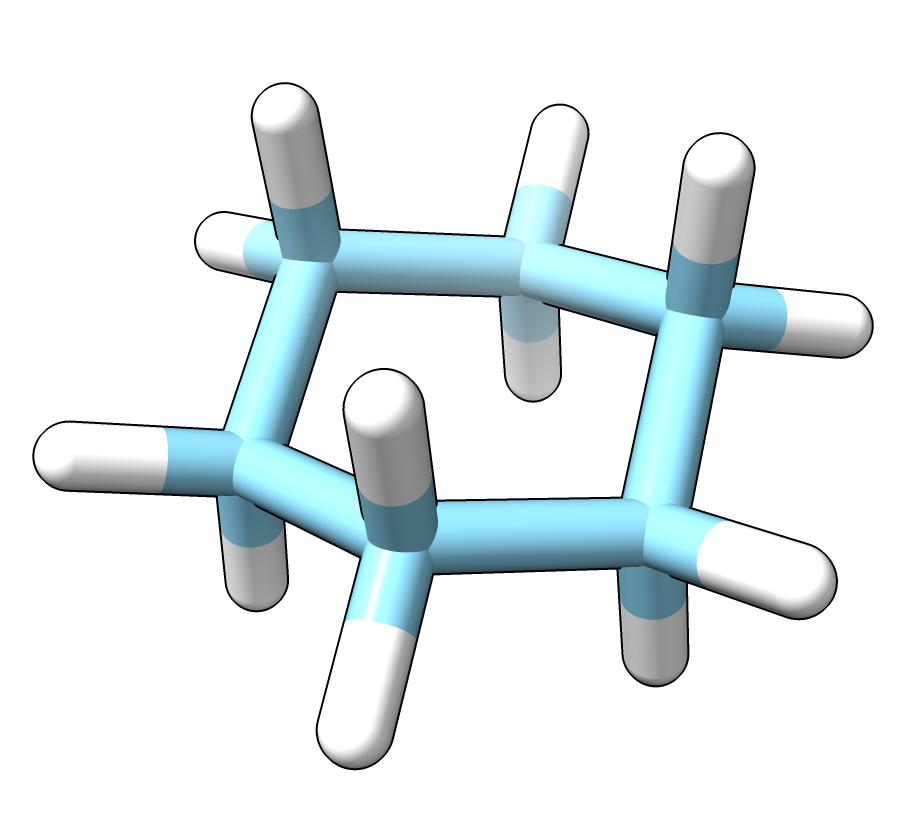
Cyclohexane predicted by AlphaFold 3
comes out as benzene (planar).
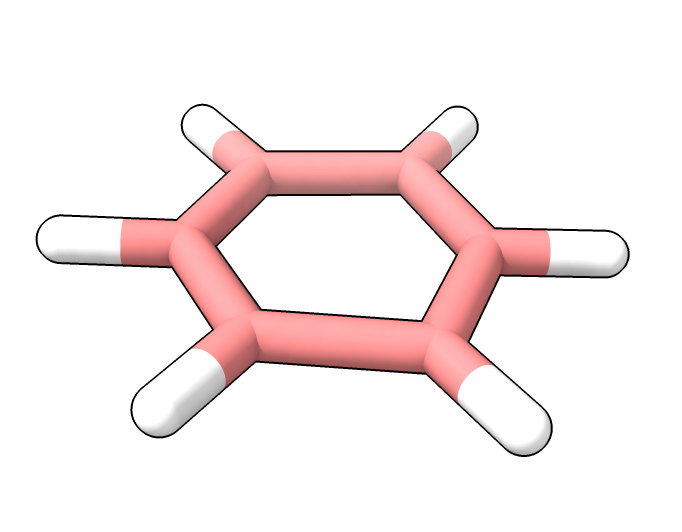
AlphaFold 3 prediction with
hydrogens added by ChimeraX
based on conformation.
Tom Goddard and Tristan Croll February 25, 2026
Structure prediction programs AlphaFold 3, OpenFold 3 and Boltz 2 appear to have little understanding of ligand chemistry, frequently predicting conformations that do not match the bond order and chirality of the input ligands. Predicted conformations would have different hydrogens and binding properties. These errors are numerous (> 50% of ligands) and likely significantly degrade the quality of ligand binding predictions.
AlphaFold 3 does not appear to understand single versus double bonds, for instance predicting a tetraheral cyclohexane ring of 6 carbons as a planar benzene ring. The predicted conformation is a different molecule with different hydrogens. Binding predictions won't be accurate if AlphaFold 3 does not understand how many hydrogens are bonded.
|
Cyclohexane (PubChem 8078). |
Cyclohexane predicted by AlphaFold 3 comes out as benzene (planar). |
AlphaFold 3 prediction with hydrogens added by ChimeraX based on conformation. |
Here we predict serotonin and 3 serotonin-like ligands varying in bond orders specified by SMILES strings, bound to a malaria protein (PDB 2qeh, not shown). Alphafold ignores the differing bond orders and predicts similar conformations for all variants.
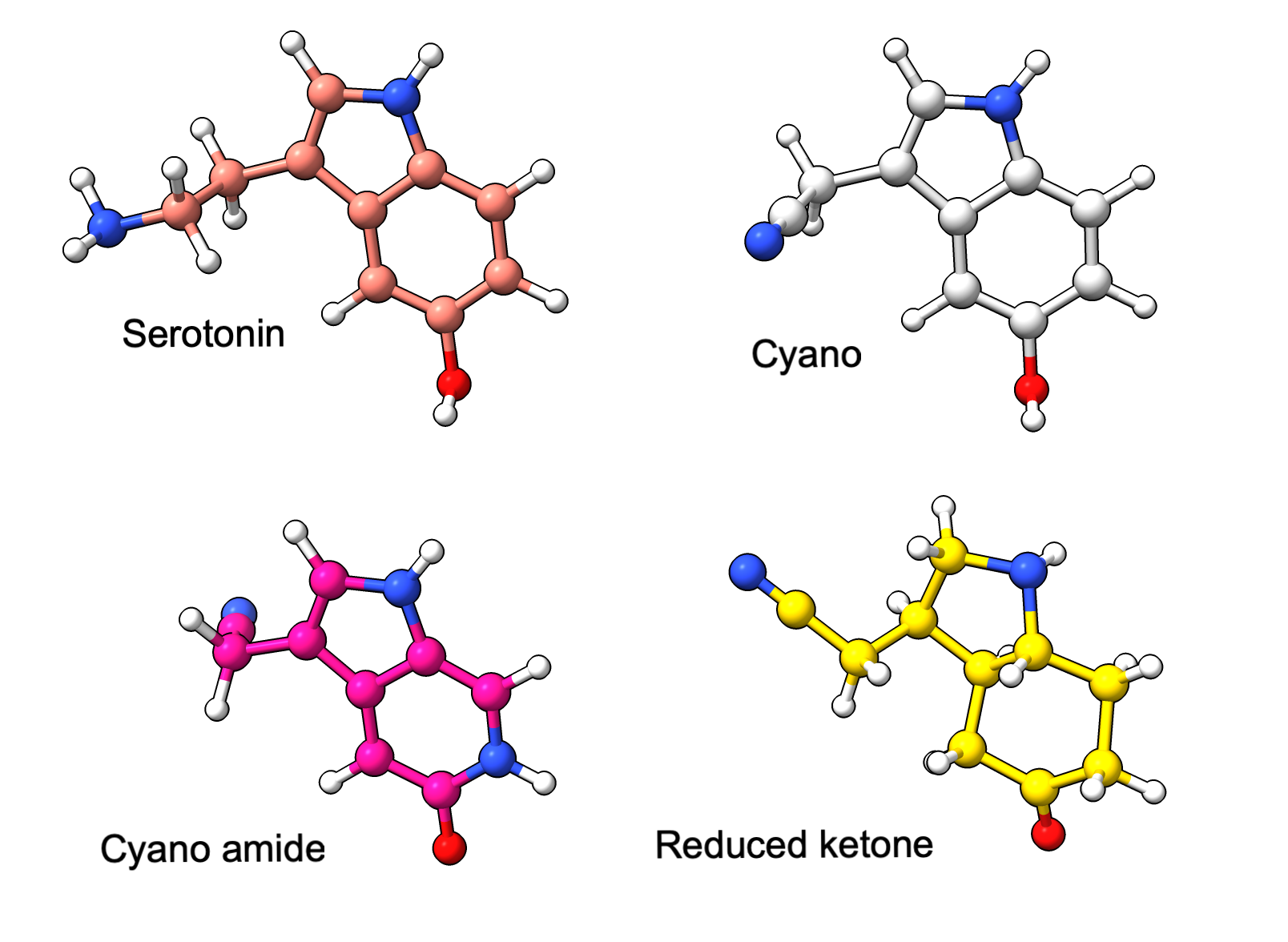
Serotonin variants. Reference conformations from SMILES. | 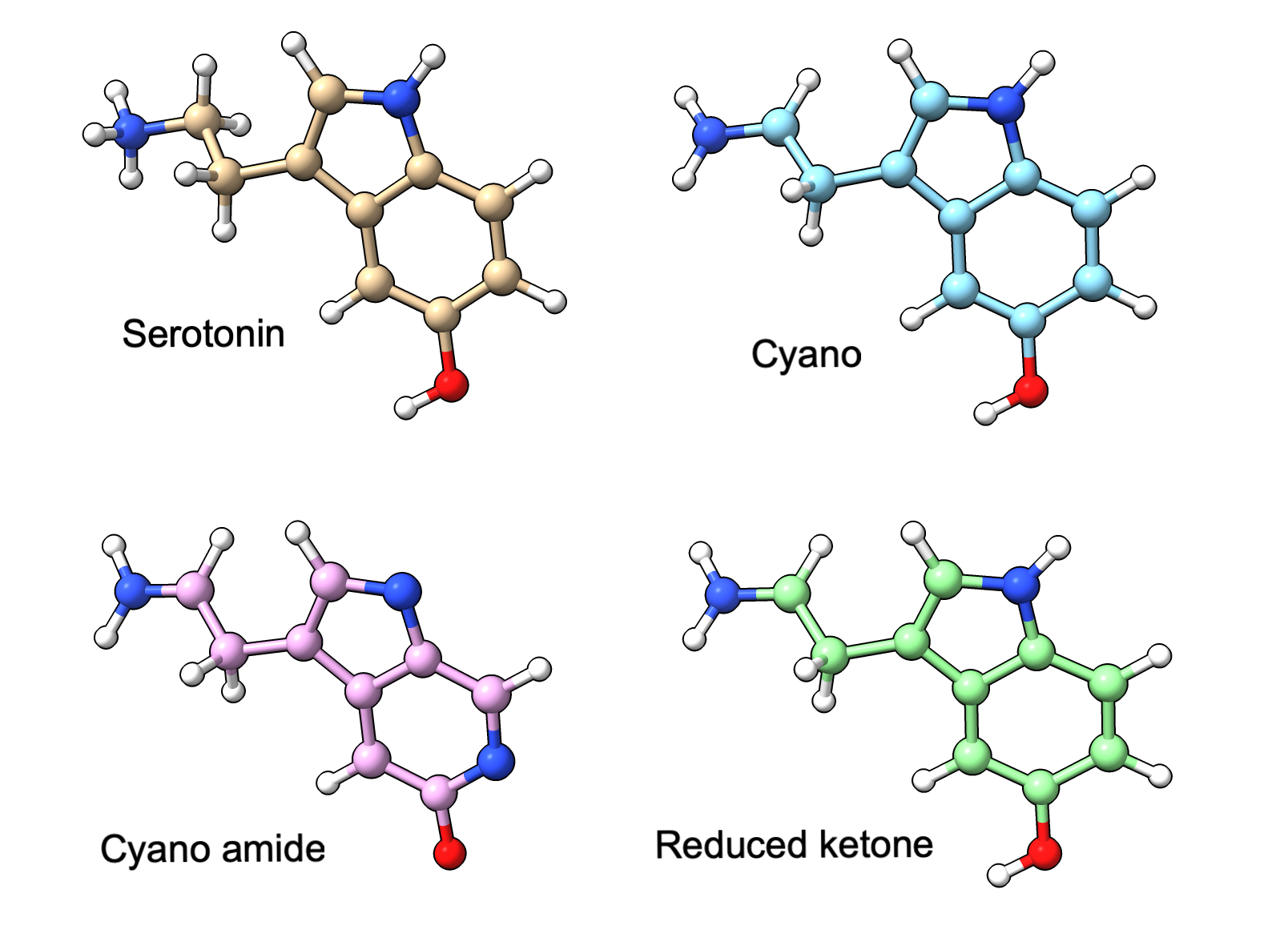
Serotonin variants predicted by AlphaFold 3 hydrogens inferred from conformations using ChimeraX. | 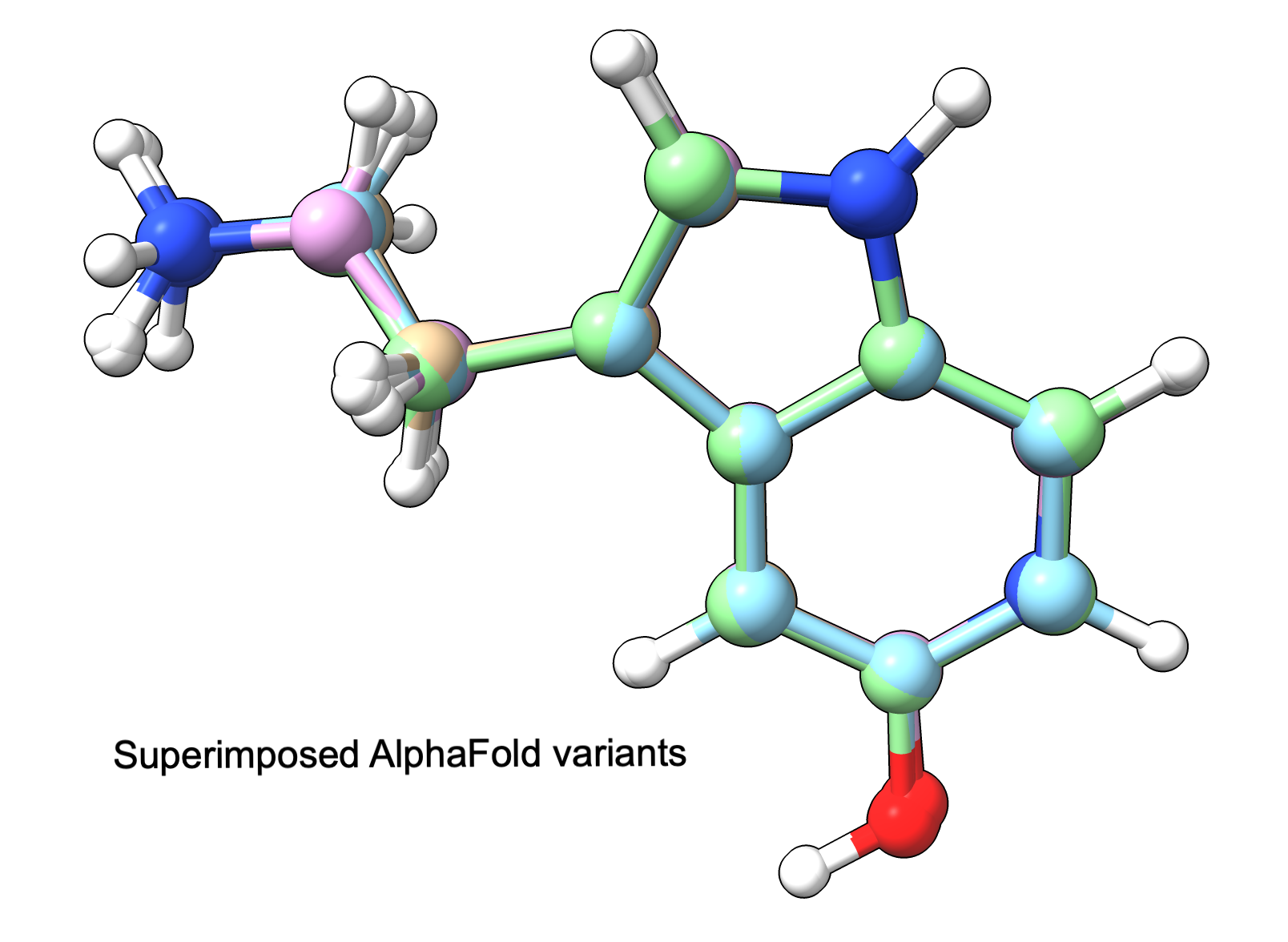
AlphaFold 3 predictions for variants all have similar conformation |
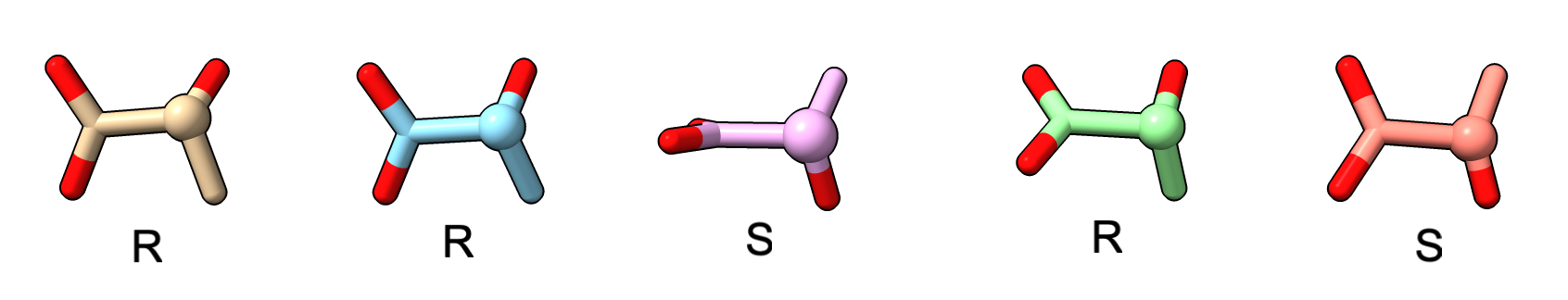
Alphafold 3 predictions frequently have the wrong chirality for ligand chiral centers. For example, here is S-lactic acid (biologically relevant form) predicted using SMILES string C[C@@H](C(=O)O)O which specifies stereochemistry. 3 of 5 of the predictions have the wrong chirality.
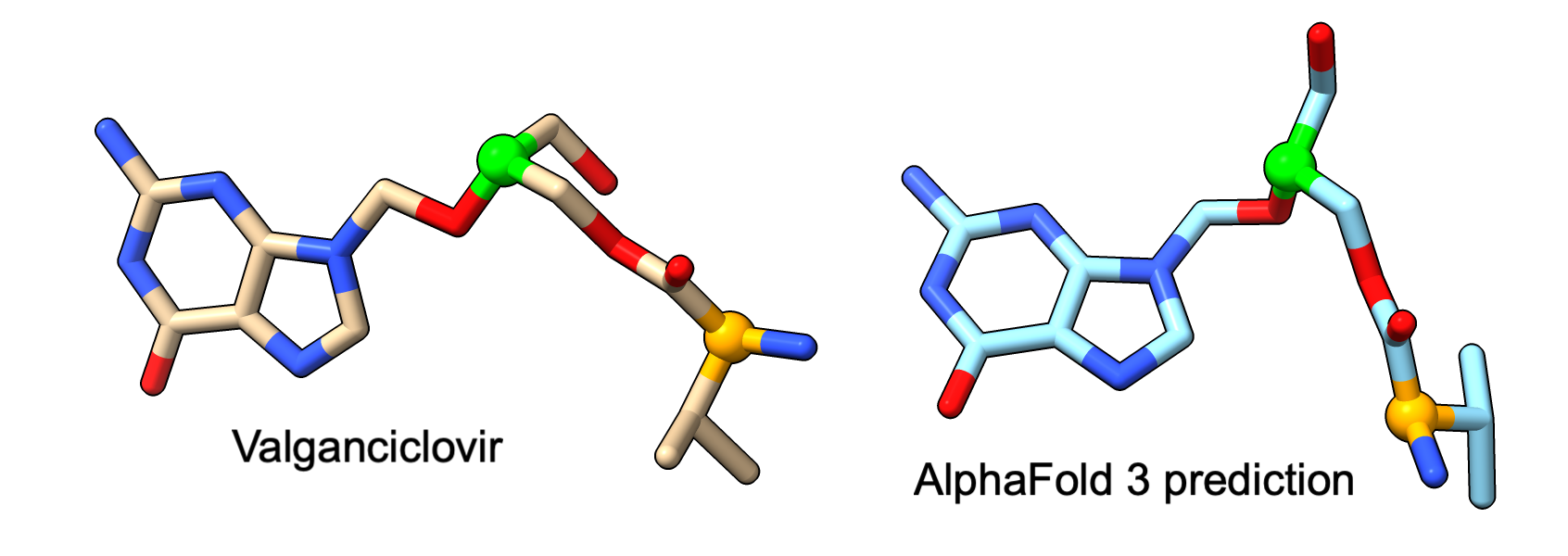
Below is another example with antiviral drug valganciclovir which has two chiral centers. AlphaFold 3 predicts one correctly (green) and one flipped (orange).
AlphaFold 3 (Mar 2026), OpenFold 3 preview 1 (Oct 2025) and preview 2 (March 2026), and Boltz 2 (Aug 2025) all produce bad ligand chemistry and chirality in predictions for 52 antiviral drugs bound to HIV protease.
The table below shows number of ligands (atoms) with chemistry and chirality errors in predictions of 52 antiviral ligands bound to HIV protease dimer. Incorrect chemistry is assessed by adding hydrogen atoms inferred from the predicted ligand conformation using the ChimeraX addh command and comparing the hydrogens added to a reference ligand conformation.
More than half of the ligands have wrong chemistry or chirality in each of the tested programs.
| AlphaFold | OpenFold preview 1 | OpenFold preview 2 | Boltz | Boltz Steering | |
|---|---|---|---|---|---|
| Ligands | 52 | 52 | 51 | 52 | 50 |
| Wrong H | 22 (73) | 27 (87) | 22 (70) | 16 (37) | 9 (29) |
| Flipped chirals | 27 (57) | 25 (47) | 16 (24) | 27 (57) | 3 (3) |
| Wrong bonds | 2 | 0 | 1 | 0 | 2 |
The above summary table shows Boltz with steering potentials enabled produces significantly fewer chemistry and chirality errors. The steering potentials apply forces to the atoms during the atom diffusion step of prediction. They do not affect the earlier PairFormer stage which infers atomic interactions. We suspect that the steering potentials fix the problems too late in the process to improve binding poses and affinity predictions. They essentially fix the geometry after the ligand placement was determined without knowledge of the hydrogens and chiralities.
Here are speculative ideas about how to improve the ligand prediction understanding of chemistry and chirality.
To test whether their are difficulties in including hydrogens on ligands in structure predictions I modified OpenFold 3 preview 1. I added a line of code to add hydrogens to the SMILES strings and another line to prevent removing hydrogens later (code diff). With those two changes OpenFold predicted cyclohexane with correct hydrogens. Unsurprisingly it did not produce any better geometry for the carbon ring, flattening it in the same was as without hydrogens. Since OpenFold was not trained with hydrogens it likely knows nothing about how they affect the structure.
Cyclohexane predicted by OpenFold including hydrogens. Ring flattened. Colored by pLDDT. |
Cyclohexane from PubChem. |
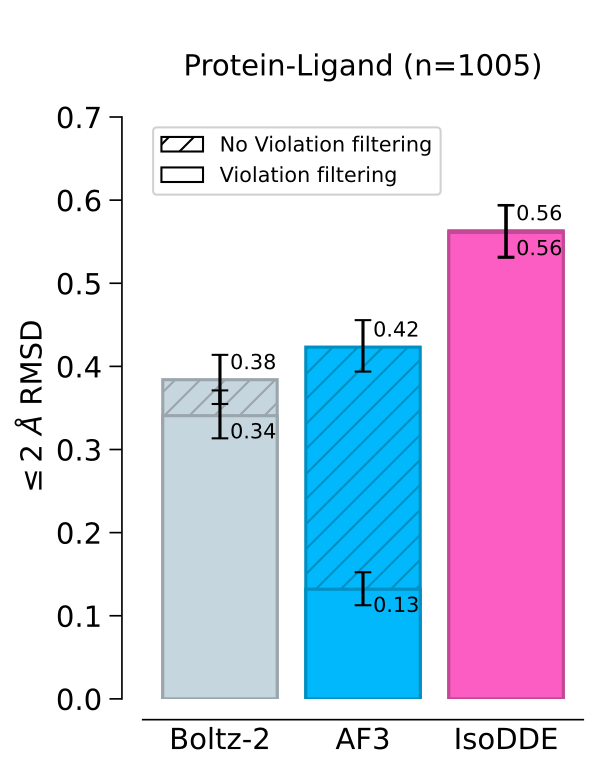
Almost all machine learning structure prediction is being pursued by companies and kept secret. They are interested in drug discovery so it seems likely they have all worked on these ligand prediction problems. Figure 5 (shown at right) from the Isomorphic Labs Drug Discovery Engine (IsoDDE) white-paper released February 10, 2026 shows AlphaFold 3 ligand binding success rate dropped from 0.42 to 0.13 if "violation filtering" was done (removing results with bad bond lengths, bond angles, wrong chiralities, clashes), while their newer IsoDDE program appears to not produce any violations. The Boltz 2 violations are relatively small (0.38 to 0.34) possibly because they enabled steering potential to fix chiralities and because their bond length/angle violation thresholds were very large. The white-paper does not give any clues how violations were reduced. The white-paper only provides benchmark results.
| wrong H | flipped chirals | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ligand | heavy atoms | H | rings | heavy atoms with H | alphafold3 | openfold3 preview 1 | openfold3 preview 2 | boltz2 | boltz2 steering | chiral centers | alphafold3 | openfold3 preview 1 | openfold3 preview 2 | boltz2 | boltz2 steering |
| abacavir sulfate | 47 | 36 | 6 | 26 | 12 C1 N1 C2 C3 N7 N9 C12 C13 C15 C16 C26 C27 | 8 N1 N2 N7 N8 C12 C13 C26 C27 | 12 C1 N1 C2 N3 N7 N8 C12 C13 C15 C16 C26 C27 | 4 C1 C2 C15 C16 | 8 C1 C2 C12 C13 C15 C16 C26 C27 | 4 | 0 | 2 C9 C23 | 0 | 0 | 0 |
| acyclovir | 16 | 11 | 2 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| acyclovir sodium | 17 | 11 | 1 | 7 | 2 O3 N4 | 2 O3 N4 | 2 O3 N4 | 2 O3 N4 | 2 O3 N4 | 0 | 0 | 0 | 0 | 0 | 0 |
| adefovir dipivoxil | 34 | 32 | 2 | 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| atazanavir sulfate | 56 | 52 | 2 | 33 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| baloxavir marboxil | 40 | 23 | 6 | 16 | 1 C14 | 2 O7 C14 | 5 C7 C8 C9 C10 C14 | 0 | 0 | 2 | 2 C10 C14 | 2 C10 C14 | 2 C10 C14 | 2 C10 C14 | 0 |
| bictegravir sodium | 33 | 18 | 4 | 13 | 3 N3 O4 C15 | 3 O2 N3 C15 | 1 O4 | 2 N3 C15 | 0 | 3 | 0 | 0 | 0 | 0 | 0 |
| brincidofovir | 38 | 51 | 1 | 27 | 2 C14 C15 | 0 | 15 C3 C4 C5 C6 C7 C8 C9 C10 C11 C12 C13 C15 C16 C17 C18 | 0 | 0 | 1 | 1 C21 | 0 | 0 | 1 C21 | 0 |
| cabotegravir sodium | 30 | 17 | 3 | 12 | 2 N3 C13 | 4 N3 C4 C11 C13 | 1 O5 | 0 | 0 | 2 | 1 C4 | 1 C4 | 0 | 1 C4 | 0 |
| cidofovir | 18 | 12 | 1 | 8 | 0 | 0 | 0 | 0 | 0 | 1 | 1 C6 | 0 | 0 | 1 C6 | 0 |
| darunavir | 38 | 37 | 4 | 26 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| darunavir dihydrate | 40 | 37 | 2 | 26 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| darunavir ethanolate | 41 | 43 | 3 | 29 | 2 C1 C2 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| darunavir hydrate | 39 | 37 | 3 | 26 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| darunavir propylene glycolate | 43 | 45 | 3 | 31 | 0 | 2 O9 C29 | 0 | 0 | 0 | 5 | 1 C29 | 1 C29 | 1 C29 | 0 | 1 C29 |
| dolutegravir sodium | 31 | 19 | 3 | 13 | 0 | 0 | 1 N3 | 0 | 0 | 2 | 2 C2 C5 | 2 C2 C5 | 0 | 2 C2 C5 | 0 |
| elbasvir | 65 | 55 | 9 | 37 | 4 N2 N3 N5 N6 | 4 N2 N3 N5 N6 | 7 C2 C4 N8 C36 C37 C38 C39 | 2 N5 N6 | 0 | 5 | 2 C4 C36 | 2 C4 C36 | 2 C4 C36 | 1 C19 | 0 |
| elvitegravir | 31 | 22 | 3 | 14 | 0 | 0 | 1 O2 | 0 | 0 | 1 | 0 | 0 | 1 C4 | 0 | 0 |
| entecavir | 20 | 15 | 3 | 11 | 0 | 0 | 0 | 2 N3 N4 | 0 | 3 | 0 | 0 | 0 | 3 C3 C5 C6 | 0 |
| entecavir anhydrous | 20 | 15 | 3 | 11 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 3 C3 C5 C6 | 0 |
| famciclovir | 23 | 19 | 2 | 10 | 2 C5 C6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| fosamprenavir calcium | 40 | 34 | 2 | 23 | 0 | 2 N2 C7 | 2 N2 C7 | 2 C5 C6 | 0 | 3 | 1 C16 | 2 C6 C7 | 1 C7 | 3 C6 C7 C16 | 0 |
| fostemsavir tromethamine | 49 | 36 | 4 | 22 | 0 | 1 O1 | 0 | 4 C13 C14 C15 C16 | 4 C13 C14 C15 C16 | 0 | 0 | 0 | 0 | 0 | 0 |
| ganciclovir | 18 | 13 | 2 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ganciclovir sodium | 19 | 13 | 1 | 9 | 2 N4 O4 | 2 N4 O4 | 2 N4 O4 | 2 N4 O4 | 2 N3 O4 | 0 | 0 | 0 | 0 | 0 | 0 |
| glecaprevir | 58 | 46 | 7 | 29 | 4 C3 C4 C24 C25 | 3 N4 C24 C25 | 2 C23 C25 | 1 C23 | 1 N4 | 7 | 3 C6 C8 C22 | 1 C8 | 1 C6 | 5 C8 C11 C13 C16 C18 | 0 |
| grazoprevir anhydrous | 54 | 50 | 7 | 30 | 4 C15 C16 C19 C20 | 3 N6 C15 C16 | 2 C15 C16 | 3 C18 C19 C20 | 0 | 7 | 3 C12 C14 C37 | 2 C35 C37 | 0 | 5 C5 C8 C14 C35 C37 | 0 |
| ledipasvir | 65 | 54 | 10 | 35 | 4 N2 N3 N4 N5 | 6 N2 N3 N4 N5 C8 C9 | 0 | 0 | 0 | 6 | 2 C36 C39 | 4 C11 C35 C36 C39 | 1 C4 | 2 C36 C39 | 0 |
| lenacapavir sodium | 65 | 32 | 6 | 20 | 4 N2 C4 N4 C5 | 3 N2 N4 C30 | 2 N2 N4 | 2 N2 N4 | 2 N2 N4 | 3 | 1 C21 | 3 C21 C33 C35 | 0 | 3 C21 C33 C35 | 0 |
| letermovir | 41 | 27 | 5 | 18 | 0 | 0 | 1 C9 | 0 | 0 | 1 | 0 | 1 C9 | 1 C9 | 1 C9 | 0 |
| lopinavir | 46 | 48 | 4 | 33 | 0 | 2 N4 C36 | 1 N4 | 1 N4 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| maribavir | 24 | 19 | 3 | 14 | 0 | 0 | 0 | 0 | 0 | 4 | 4 C11 C12 C13 C14 | 4 C11 C12 C13 C14 | 0 | 4 C11 C12 C13 C14 | 0 |
| molnupiravir | 23 | 19 | 2 | 14 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| nelfinavir mesylate | 45 | 49 | 3 | 31 | 0 | 4 C21 C22 C23 C24 | 0 | 0 | 0 | 6 | 0 | 1 C20 | 0 | 1 C20 | 0 |
| oseltamivir phosphate | 27 | 29 | 0 | 15 | 1 C6 | 4 N2 C9 C10 C11 | 3 C4 C5 O5 | 2 N1 C11 | 0 | 3 | 3 C6 C10 C11 | 2 C10 C11 | 0 | 1 C11 | 0 |
| penciclovir | 18 | 15 | 2 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| peramivir | 23 | 28 | 1 | 17 | 0 | 3 N4 C6 C8 | 0 | 0 | 1 N4 | 5 | 5 C6 C7 C8 C10 C11 | 4 C6 C8 C10 C11 | 0 | 1 C7 | 0 |
| pibrentasvir | 80 | 65 | 10 | 41 | 15 C1 C2 N6 N7 C16 C17 C18 C19 C26 C27 C28 C29 C30 C50 C51 | 5 N2 N3 N6 N7 C19 | 2 C17 C18 | 4 N2 N3 N6 N7 | 4 N3 N6 N7 C13 | 8 | 4 C2 C16 C19 C50 | 1 C19 | 4 C2 C16 C19 C50 | 2 C16 C19 | 0 |
| raltegravir potassium | 33 | 21 | 2 | 12 | 1 O4 | 0 | 1 O4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| remdesivir | 42 | 35 | 4 | 24 | 2 C4 C5 | 2 N2 C15 | 0 | 0 | 0 | 6 | 2 P1 C8 | 1 C8 | 0 | 0 | 0 |
| ritonavir | 50 | 48 | 4 | 33 | 0 | 1 C7 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| sofosbuvir | 36 | 29 | 3 | 20 | 0 | 0 | 0 | 0 | 0 | 6 | 1 C2 | 3 P1 C2 C10 | 1 P1 | 0 | 0 |
| tenofovir alafenamide | 33 | 29 | 3 | 18 | 0 | 0 | 0 | 0 | 0 | 3 | 3 P1 C2 C10 | 2 P1 C2 | 2 P1 C2 | 3 P1 C2 C10 | 0 |
| tenofovir alafenamide fumarate | 41 | 31 | 2 | 20 | 2 C22 C23 | 2 C22 C23 | 0 | 0 | 0 | 3 | 2 C2 C10 | 1 C2 | 2 P1 C2 | 1 C2 | 0 |
| tenofovir disoproxil fumarate | 43 | 32 | 1 | 17 | 2 C20 C21 | 2 C20 C21 | 0 | 0 | 0 | 1 | 1 C2 | 1 C2 | 0 | 1 C2 | 0 |
| tipranavir | 42 | 33 | 4 | 23 | 1 O5 | 2 C24 C25 | 1 O5 | 1 O5 | 0 | 2 | 1 C4 | 2 C4 C9 | 1 C4 | 2 C4 C9 | 0 |
| valacyclovir hydrochloride | 24 | 21 | 1 | 11 | 0 | 0 | 0 | 0 | 0 | 1 | 1 C4 | 0 | 0 | 0 | 0 |
| valganciclovir | 25 | 23 | 2 | 13 | 0 | 0 | 0 | 0 | 0 | 2 | 1 C4 | 0 | 0 | 1 C4 | 1 C7 |
| valganciclovir hydrochloride | 26 | 23 | 1 | 13 | 0 | 0 | 0 | 0 | 0 | 2 | 1 C7 | 0 | 1 C7 | 1 C7 | 1 C7 |
| velpatasvir | 65 | 54 | 9 | 36 | 1 C24 | 6 N5 N6 C8 N8 C10 C24 | 4 C5 N5 N6 C24 | 3 N5 N6 C24 | 5 N3 N4 N5 C24 C33 | 6 | 5 C2 C5 C7 C34 C39 | 1 C39 | 2 C2 C5 | 3 C7 C34 C36 | 0 |
| voxilaprevir | 60 | 52 | 7 | 30 | 0 | 7 C19 C20 N26 C32 C43 C44 C45 | 2 C32 C45 | 0 | 0 | 8 | 3 C05 C09 C17 | 1 C09 | 0 | 3 C05 C17 C31 | 0 |
| zanamivir | 23 | 20 | 1 | 15 | 0 | 2 C5 C6 | 0 | 0 | 0 | 5 | 0 | 0 | 1 C9 | 0 | 0 |
Incorrect bonds. Two of 52 boltz steering predictions contained bonds not present in the input. This came about because the mmCIF atomic model output written by all the programs does not contain bond information for the ligands. Therefore the bonds must be guessed based on atom positions. With Boltz steering an oxygen of a ring structure was moved 5 Angstroms away from its correct ring position. The bad bonds were in Boltz steering predictions for tipranavir, ledipasvir. We did not analyze chemistry and chirality mistakes for the ligands with incorrect bonds. The mmCIF output should contain explicit bond information for the ligands given that the programs are prone to atom steric clashes.
Incorrect atoms. Boltz writes mmCIF predictions with sodium atoms (Na) annotated as nitrogen and calcium atoms (Ca) annotated as carbon. This threw off comparisons that expected the same ligand atomic elements. I patched it in the comparison scripts. Ideally this would be fixed in Boltz code, although the open source Boltz project appears to have ended with the development team starting a company.
Chirality definition. Predicting incorrect chemistry leads to different placement and numbers of hydrogen atoms. That can change the ordering of the 4 substituents of a chiral center making it appear that the chirality changed when in fact the problem is a change in hydrogens. To avoid flagging those problems as chirality errors I measure chirality simply by the placement of the 4 neighbor atoms (ordered by atom name) when comparing reference ligand conformations to predicted conformations.
SMILES to 3d fails. To produce reference conformations for the 52 antiviral smiles strings I used the ChimeraX SMILES to 3d service hosted by the National Cancer Institute. This can fail to produce a 3D conformation, and did fail for the ligand voxilaprevir. I got a reference 3D conformation for voxilaprevir from PubChem. AlphaFold, OpenFold and Boltz all use RDKit to get 3D conformations from SMILES strings to use in the input features. All will use "random" coordinates from RDKit if it fails to produce good geometry. That can fail also (not sure how) in which case Boltz and OpenFold fail to predict, but AlphaFold 3 sets all atom coordinates to 0. I believe the only way the input features indicate ligand chemistry and chirality is via the input coordinates. So if the coordinate calculation is bad, this will obviously prevent the programs from producing the desired chemistry and chirality.
The results shown above can be downloaded: benzene.zip, serotonin.zip, lactic_acid.zip, antivirals.zip. A ChimeraX command script ligchem.py defining ChimeraX commands ligdiff and matchnames was used to look for chemistry and chirality differences between reference and predicted ligands.