Contact PAE color scale.
PAE heatmap color scale.
Tom Goddard
September 15, 2025, updated Sept 24, 2025
Can protein-protein interactions be predicted on inexpensive desktop computers? Is it is feasible to compute thousands of protein dimer structures using machine learning structure prediction to determine which proteins bind to each other without AI GPUs, computing clusters, or cloud computing resources that many biology researchers lack?
I ran about 4600 predictions of protein pairs in pathogenic bacterium Mycoplasma genitalium which has a minimal genome consisting of only 476 genes. Interacting proteins are likely to be positioned close together on the genome and co-expressed, so I predicted only protein pairs within 10 genes of each other on the genome. There are 5181 pairs (including homodimers) spaced at most 10 genes apart and I predict 4598 omitting 583 that are over 1200 residues in length to avoid running out of GPU memory.
Contact PAE color scale. |
PAE heatmap color scale. |
The predictions took about 30 hours on a desktop computer.
Boltz reports several confidence scores including interface predicted template model score ipTM which is the most frequently used to assess confidence in the protein-protein binding interface.
|
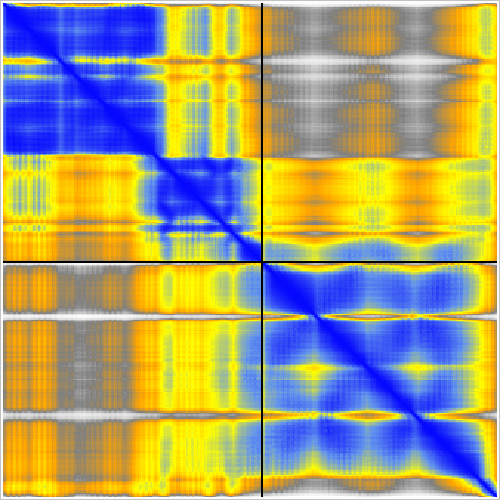
Example dimer predictions. Contacts within 5A colored by predicted aligned error (PAE). | ||
|---|---|---|
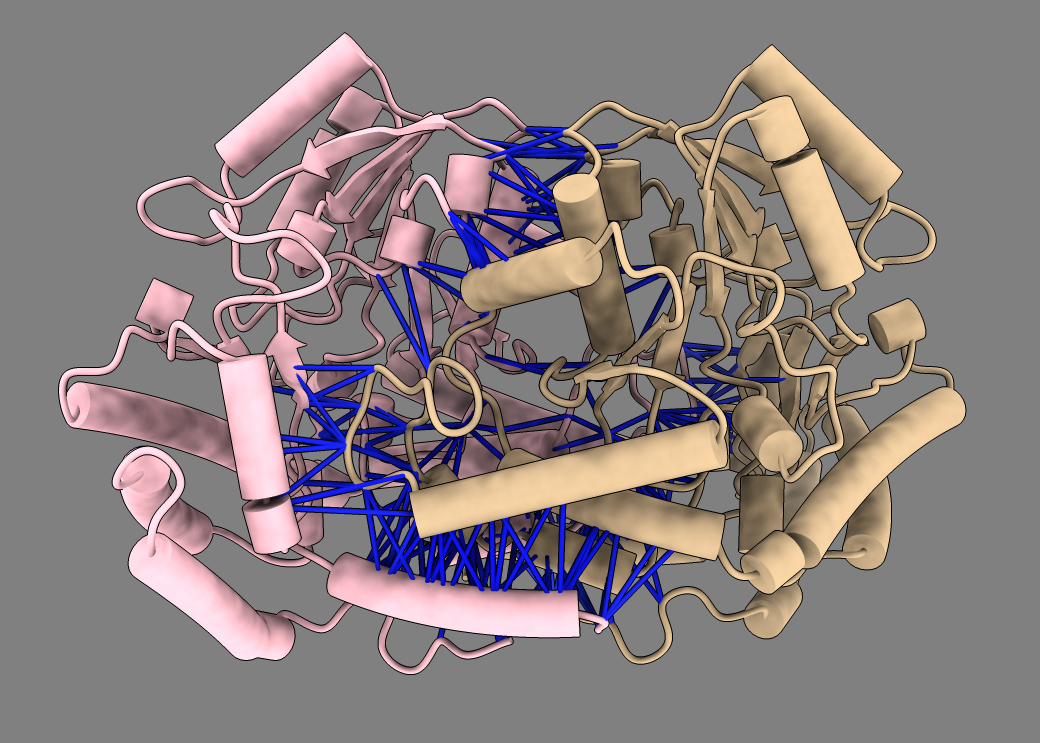
MG_394_glyA homodimer ipTM 0.98 | 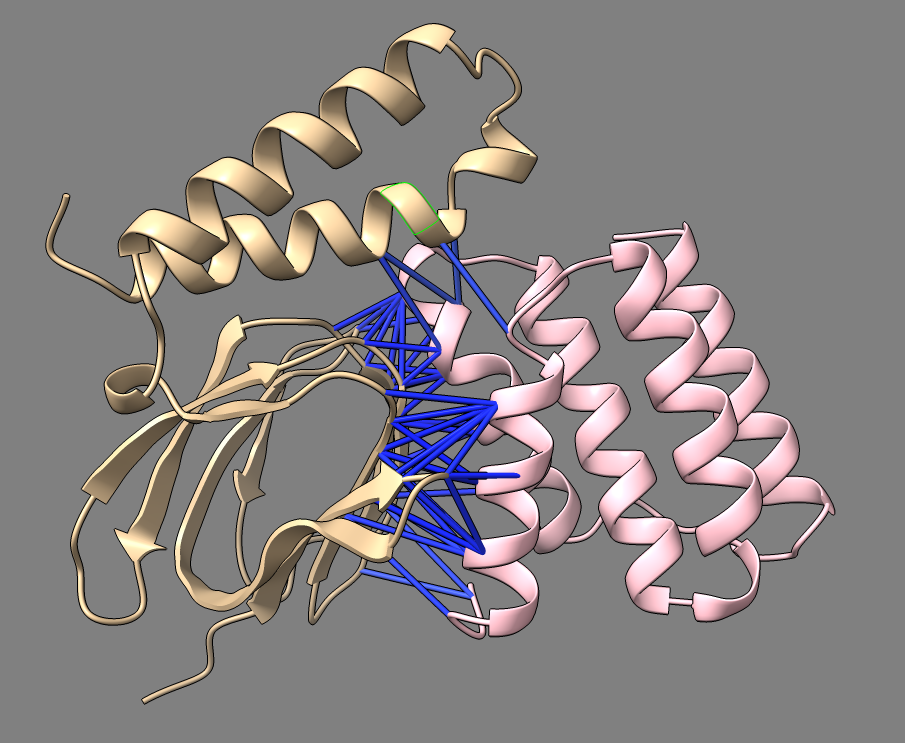
MG_398_atpC and MG_404_atpE ipTM 0.90 | 
MG_172_map and MG_176_rpsK ipTM 0.80 |
Predicted aligned error | 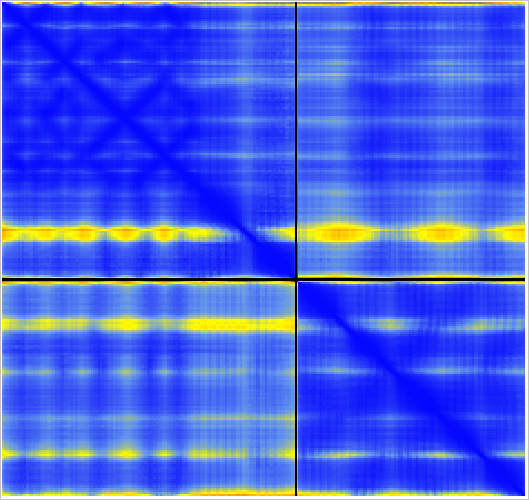
| 
|
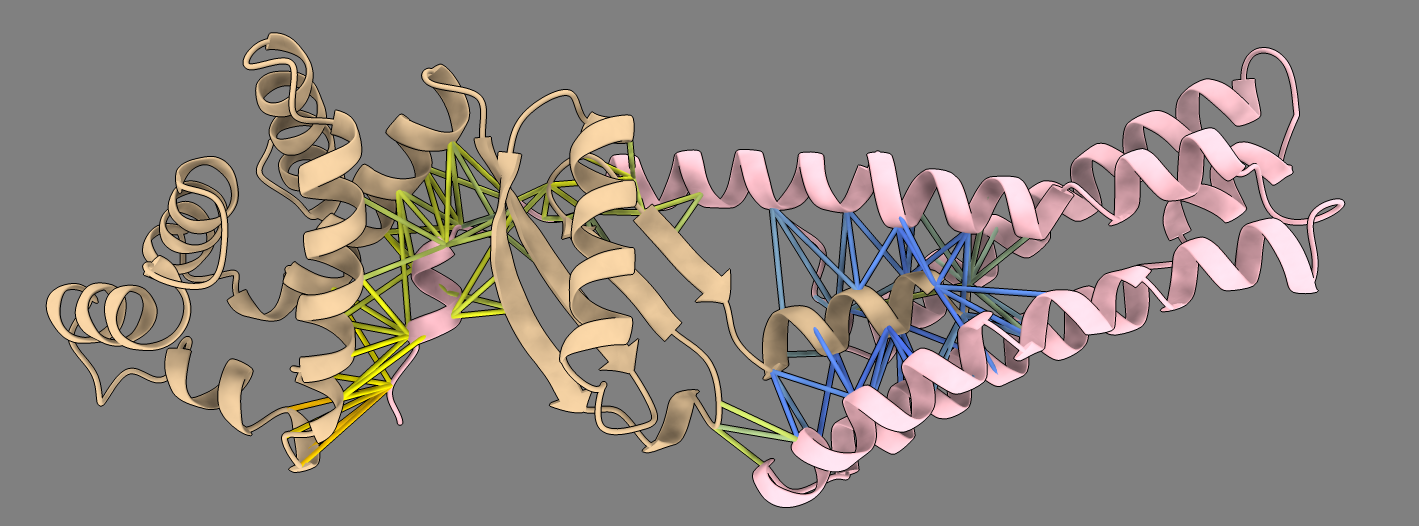
MG_402_atpH and MG_406 ipTM 0.70 | 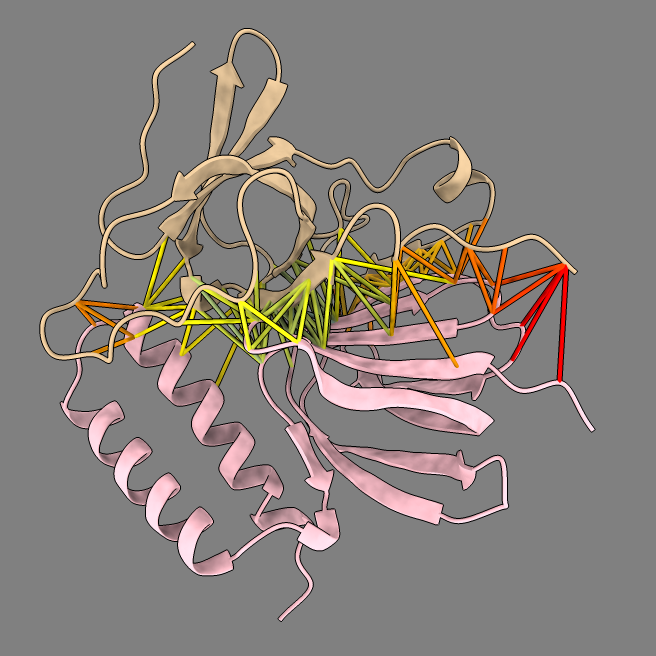
MG_393_groES and MG_398_atpC ipTM 0.60 | 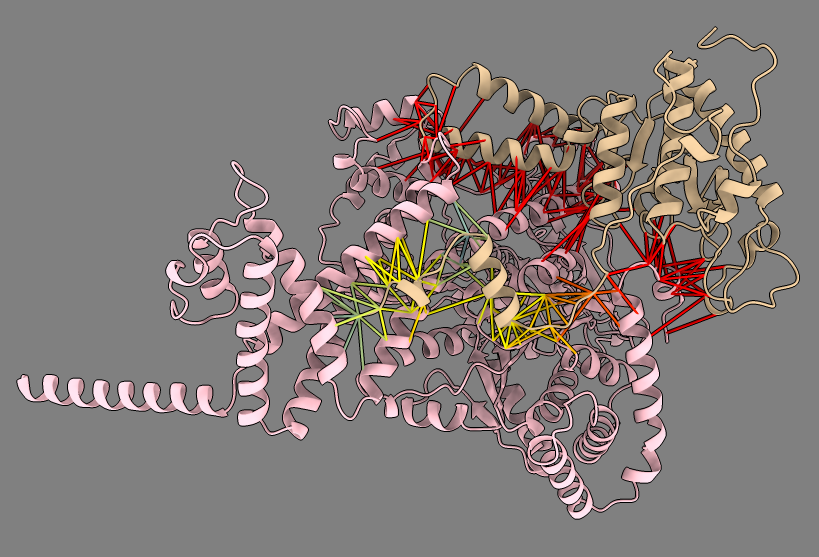
MG_070_rpsB and MG_072_secA ipTM 0.50 |
Predicted aligned error | 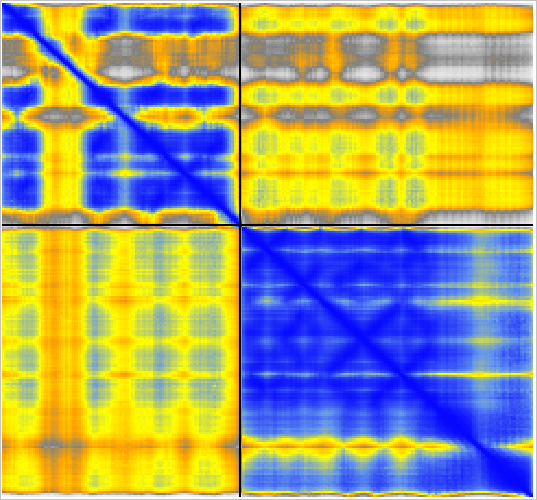
| 
|
Below are the number of pairs with ipTM greater than a given value. EBI says scores greater than 0.8 are considered confident and 0.6-0.8 gray area. All of the 4598 Boltz predictions completed. There were no "out of memory" errors that can occur if the structure has too many residues or the multiple sequence alignment contains too many sequences for the available GPU memory.
ipTM N homodimer heterodimer
0.9 65 55 10
0.8 155 90 65
0.7 287 127 160
0.6 484 157 327
0.5 781 201 580
There is a surprising lack of agreement between these Boltz 2 ipTM scores and AlphaFold 3 ipTM scores for the same protein-protein interactions computed by Horia Todor. I made a table showing the Boltz 2 and AF3 scores.
Horia predicted the protein-protein interactions in batches, pooling 15-20 proteins in a single prediction and observing which proteins stick to each other. Horia notes that pooling produces very different (better) results compared to predicting individual dimers even when both use AlphaFold 3 with a low Pearson correlation coefficient R = 0.16.
It's not yet clear how valuable large scale computational searches for protein-protein interactions. I think this is because relatively few researchers have been able to run searches because of perceived steep computer hardware requirements and software setup difficulty. My aim is to make it simple enough so more studies will reveal the utility of these computational screens.
Examples of screens. All report putative and interesting newly discovered interactions.
I used a Linux (Ubuntu 24.04) desktop computer with an Nvidia RTX 4090 graphics card with Boltz 2.2 used for structure predictions and colabfold_search and MMseqs2 used for computing paired deep sequence alignments, and custom Python scripts to create the needed input files. See the commands to install the software and run the predictions below.
Took 33 hours total: 4 hours to compute input sequence alignments and 29 hours to predict structures.
Standard MSA calculation in AlphaFold, Boltz, Chai, Colabfold repeats the database search and alignment for the same proteins each time a new combination of proteins is requested. When trying to find interactions in large sets of proteins this leads to many repeated sequence searches for each protein. In my Mycoplasma example each protein occurs in about 20 complexes (the other proteins within 10 genes along the genome), leading to 20 duplicate sequence searches for each protein. To speed this up I compute protein monomer alignments one time and make paired alignments from the monomer alignments.
| MSA | sequences | file |
|---|---|---|
| MG_007 filtered | 730 | MG_007.a3m |
| MG_007 unfiltered | 248,565 | MG_007.pre_paired_tax.a3m |
| MG_011 filtered | 330 | MG_011.a3m |
| MG_011 unfiltered | 531,867 | MG_011.pre_paired_tax.a3m |
| Paired by species | 45,007 | paired.fasta |
Look at the MSA for one Mycoplasma dimer for genes MG_007 and MG_011.
Sequence alignments with thousands of sequences can be viewed with the ChimeraX Profile Grid tool that show the percentage occurence of each amino acid at each position.

Profile grid for paired MG_007 and MG_011 MSA, 45007 sequences. Reference sequence highlighted in green. | 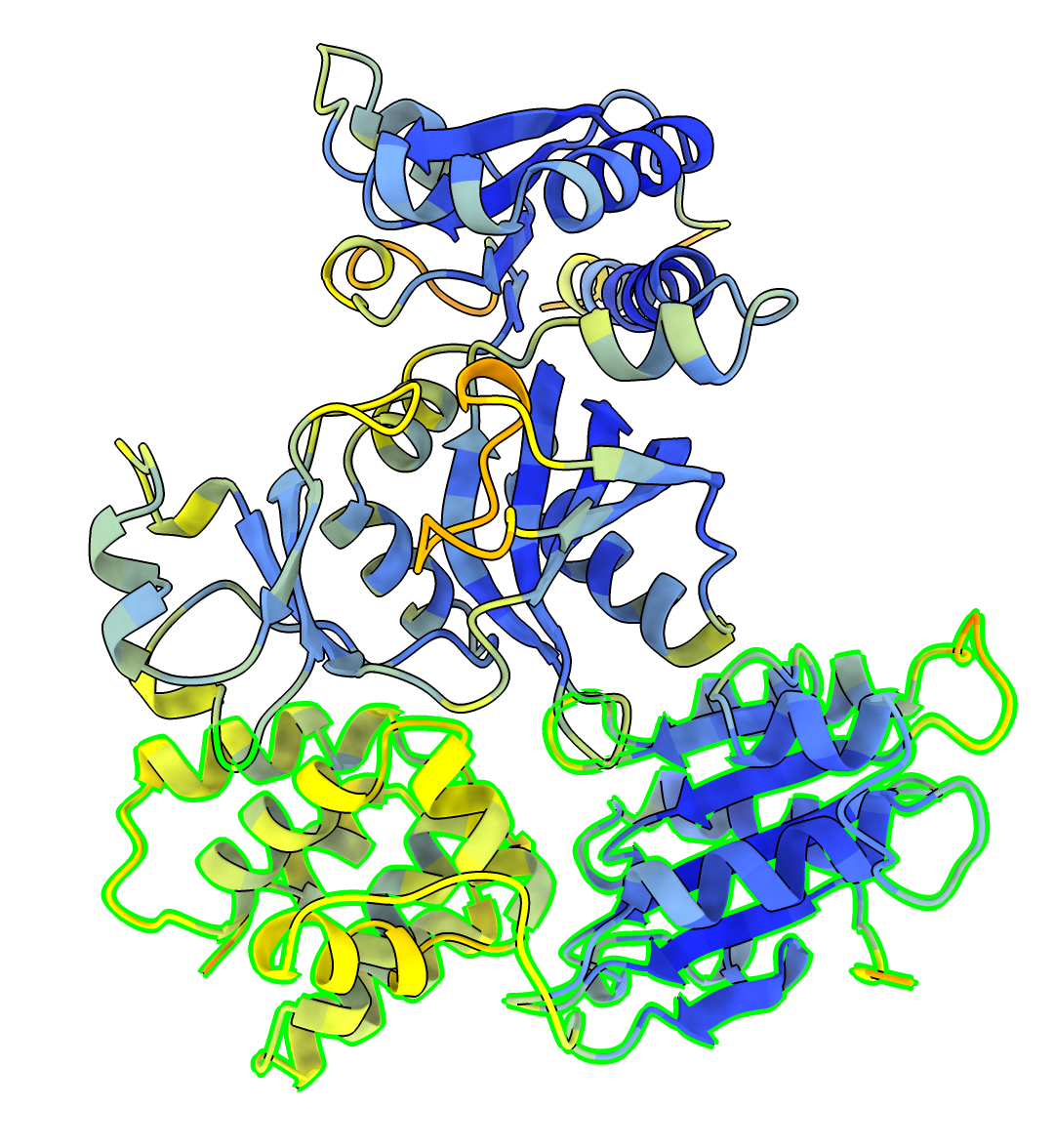
Boltz predicted dimer MG_007 and MG_011. |
A taxonomy tree of the 45007 paired sequences shows most are from bacteria (created by NCBI taxonomy common tree web page). Would be interesting to be able to filter paired MSA to chosen taxonomic groups and rerun the Boltz prediction to look for better quality preditions.

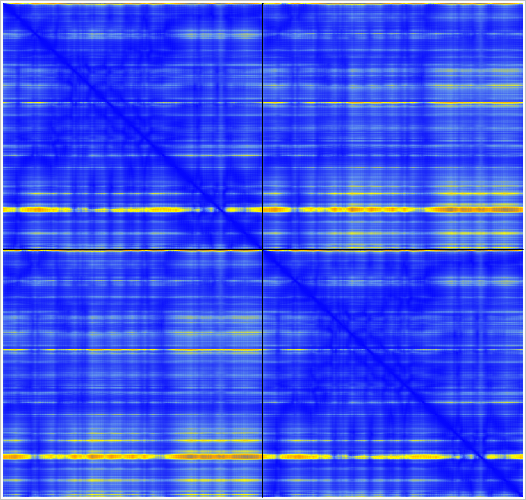
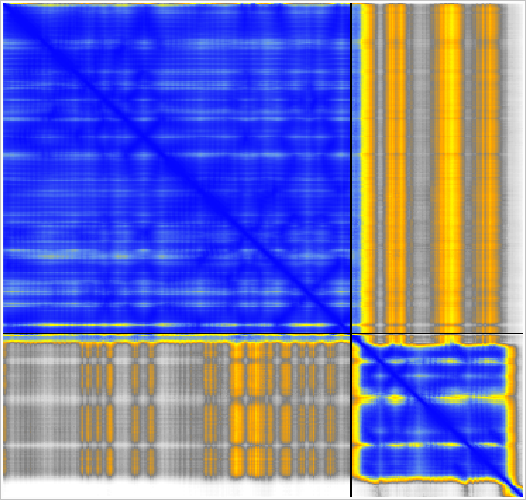
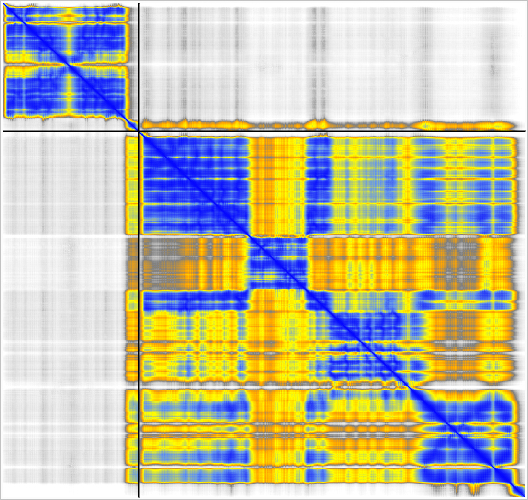
The MG_007 and MG_011 example dimer prediction gave different results with MSA computed by two slightly different methods.
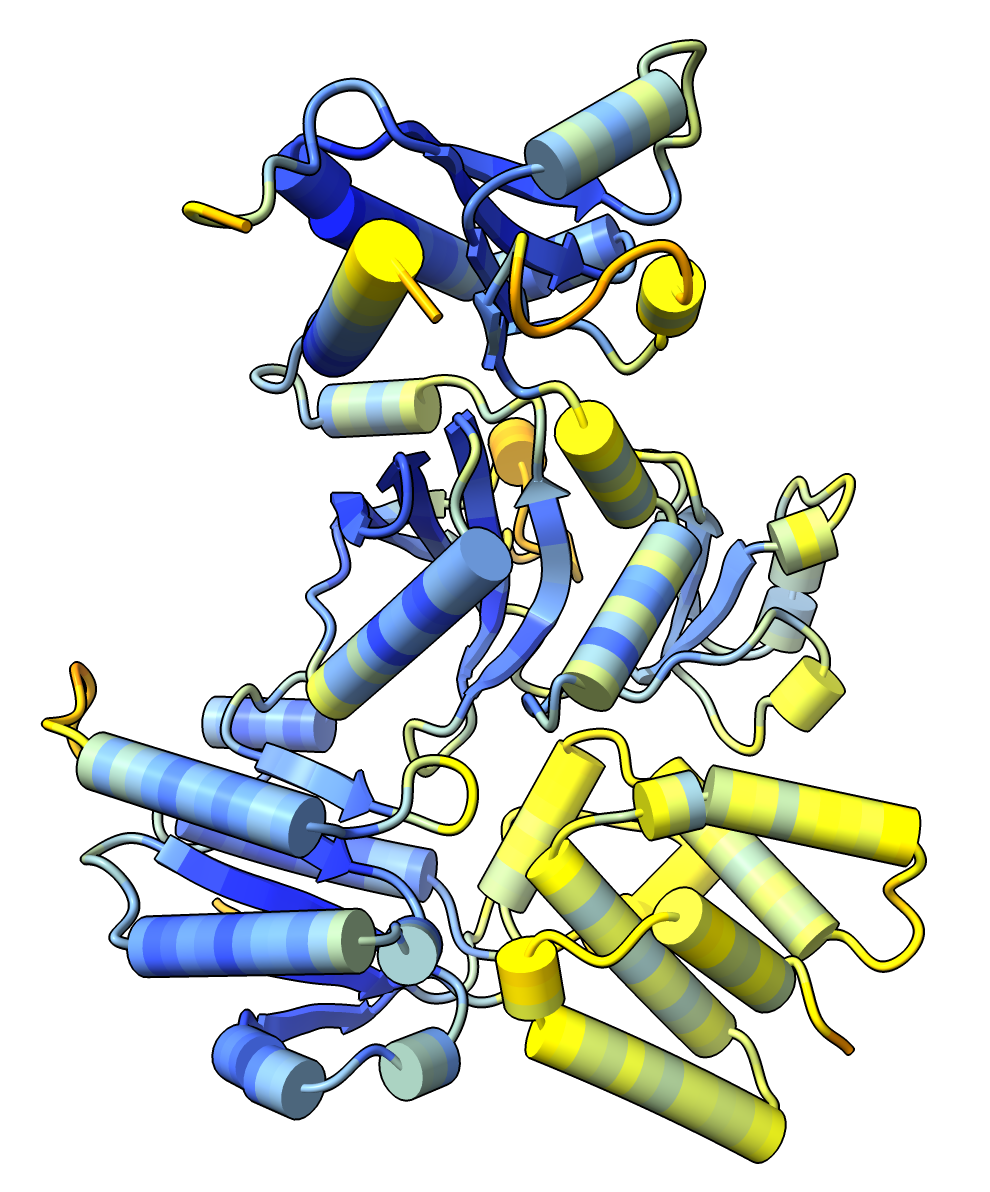
GPU ungapped prefilter. | 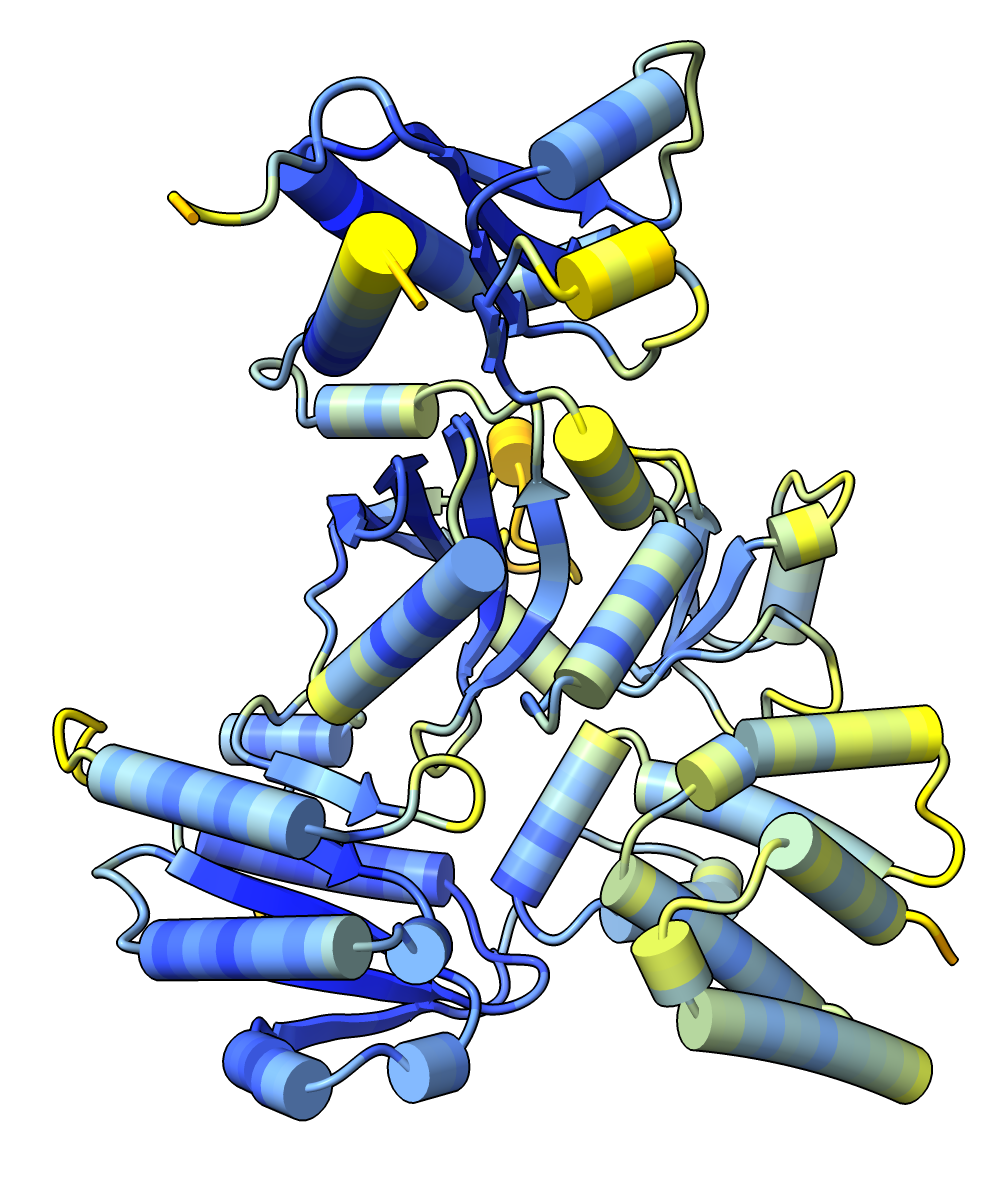
CPU kmer prefilter. | 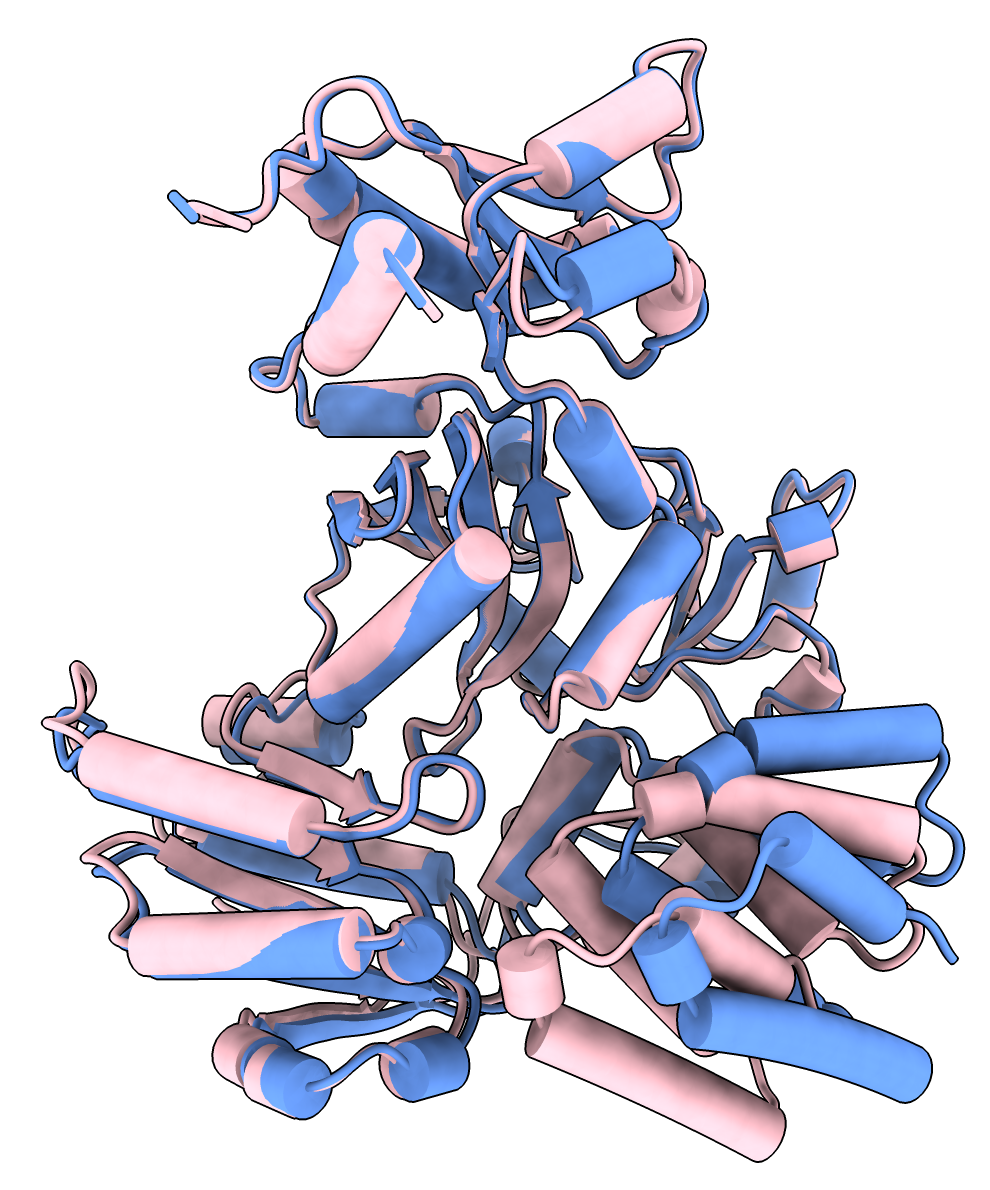
Superimposed shows alternate domain alignment (lower right). |
I ran into several problems producing and using MSAs using colabfold_search and Boltz. Here are details of the problems and fixes.
Boltz uses empty MSAs if first MSA sequence is not query. My first attempt at the dimer predictions used MSAs where the first sequences was not the query sequence, due to a mistake in my Python script that created .csv MSA files. Instead the first aligned sequence was first. Surprisingly Boltz made the predictions anyways and replaced the specified .csv MSA with an empty MSA. I only noticed this when examining Boltz log output. This caused 309 of the monomers to get empty MSAs while 167 got correct MSAs because the first aligned sequence happened to be identical to the query. Only dimers with both proteins in the correct MSA set were predicted correctly. I fixed the script and reran the predictions.
Failed predictions with large MSAs.
With correct MSAs a large fraction 25% of the Boltz predictions ran out of memory.
Investigation showed that dimers with more than 730 residues and over 8000 paired
MSA sequences almost all failed, while dimers with just hundreds of paired sequences could be predicted
up to about 1100 residues without running out of memory. So Boltz uses a good bit of memory for
the paired MSA sequences.
Reducing MSA sequence pairs to predict larger structures.
To allow more predictions to succeed I decided to reduce the maximum number of MSA paired sequences
from the Boltz default of 8192 when larger numbers of residues are predicted.
I used the following formula estimating memory use
A*n_pair*n_res + B*n_res*n_res = GPU_memory
and determined A = 19151, B = 2602 by using GPU_memory 24 * 1024 * 1024 * 1024 (24 Gbytes) and n_pair = 8192, nres = 730 and n_pair = 0, nres = 1160. For a given number of residues in a dimer I computed the n_pair value that uses 24 Gbytes, and use that as the maximum number of sequence pairs to accept in the MSA. If the computed MSA has more pairs I only take the first n_pair of them. I also limit the unpaired sequences to the same number. Since the limit n_pair can be 0 which would lead to empty MSAs and likely poor prediction quality, I use a limit of 512 paired and 512 unpaired sequences if the limit is under 512. With these limits all 4598 predictions completed.
Boltz mistakenly duplicated paired MSA sequences as unpaired.
While investigating Boltz MSA processing code to get a better idea of how much memory is used
I found two significant Boltz code errors. First it by mistake includes a copy of all paired sequences
as unpaired sequences in the MSA. The Boltz filtering code that was intended to filter out the
paired sequences was incorrect. If the MSA .csv files had 8192 paired sequences then Boltz duplicates
left no room for any of the .csv file unpaired sequences since the MSA size is capped at 16384.
Actually those caps are used when Boltz reads the MSA files. Then Boltz imposes a 8192 limit
to the total MSA number of sequences (paired plus unpaired). This can be altered with Boltz option
--max_msa_seqs (default 8192).
Boltz mistakenly removes identical MSA sequences that are paired to non-identical sequences.
Another error was when Boltz reads each protein MSA .csv it removes all duplicate sequences.
That ends up removing pairs if one protein in the pair has a sequence seen in another pair.
That can easily happen with a highly conserved protein. I was testing with Mycoplasma g. proteins
7 and 11 and this code mistake caused Boltz to toss out 2000 of the 8000 paired sequences in my .csv
MSA files. I fixed both these bugs (and reported as Boltz github issues
#581 and
#583.
The Boltz fork that ChimeraX installs was updated to include these fixes on October 3, 2025.
Fixed dimer predictions.
I reran the 4600 dimer predictions with fixed Boltz code and using a limit on the
number of paired MSA sequences that depends on residue count and this web page shows
those results (ipTM scores, dimer images, ...). I have not yet put the Boltz MSA processing
fixes into the ChimeraX Boltz installation.
| 
|
Comparison to dimer predictions without reducing MSA.
I reran the 4598 dimer predictions without reducing MSA size with the Boltz MSA fixes and
only 17 ran out of memory, all with sequence lengths 1160-1200. Rerunning those 17 as a batch
only 1 ran out of memory, and running that 1 alone succeeded. I'm not sure why the out of memory
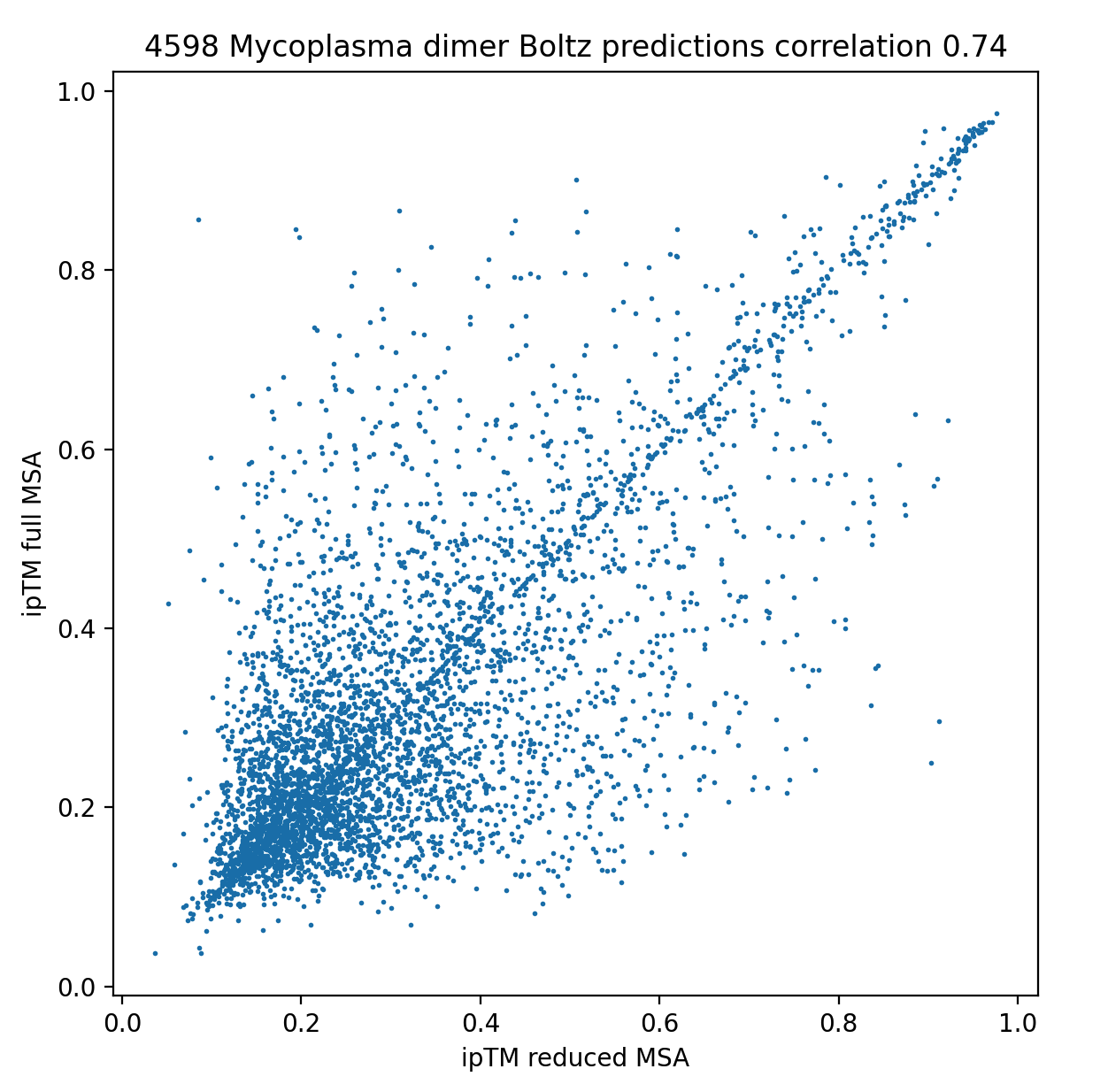
failures are not reproducible. I made a scatter plot showing ipTM scores of the dimers using
reduced MSAs versus full MSAs, correlation coefficient was 0.74. Both sets of predictions
produced only 1 structure per dimer.
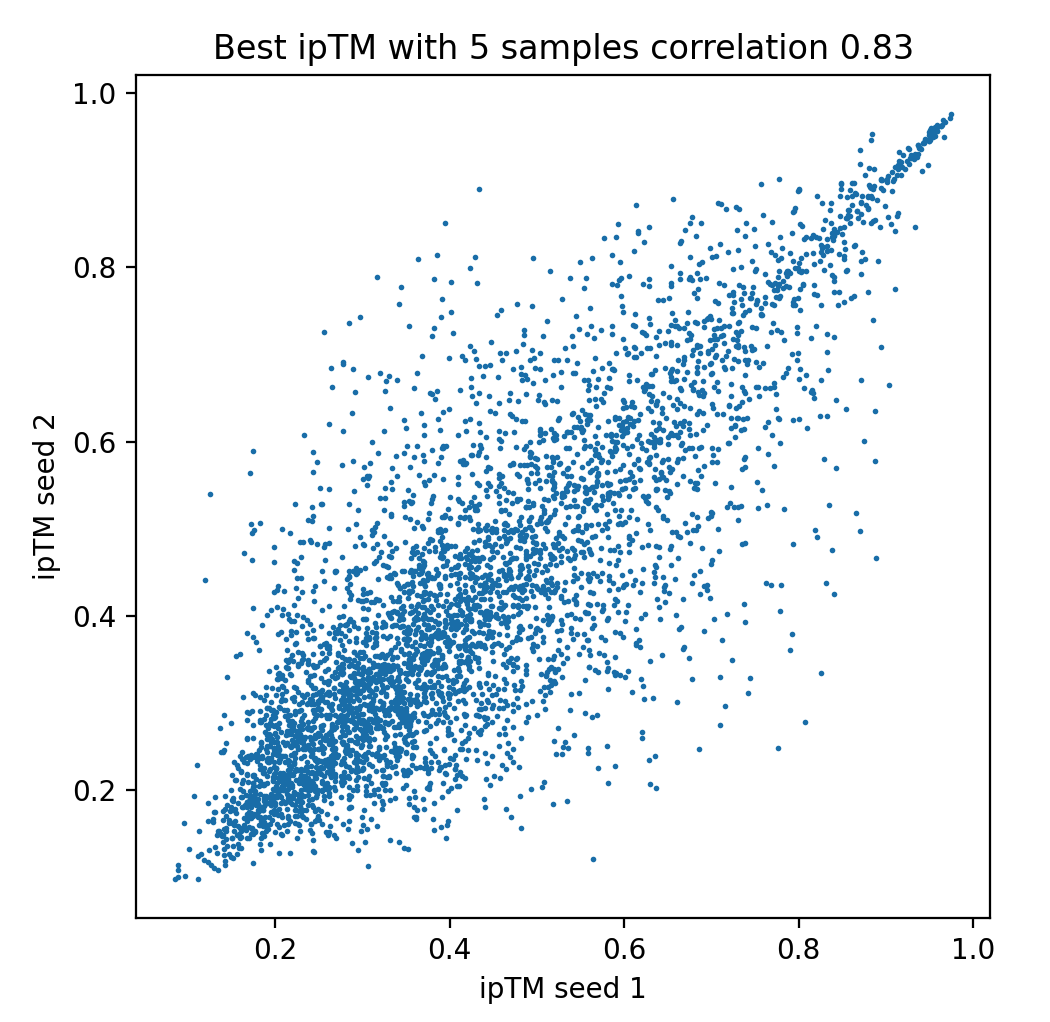
When predicting 5 structures per dimer and taking the highest ipTM using runs with random seed values 1 and 2 we get a slightly higher correlation of 0.83, but still significant variability.
It is a good idea to predict the monomer protein structures. If those cannot be predicted with good confidence scores then it is unlikely that dimers will predict correctly.
Use ChimeraX alphafold monomers command (in September 18, 2025 daily build or newer) to create boltz input files that use precomputed MSAs. Computing 466 monomers with lengths less than 1200 residues took 73 minutes. 418 have predicted template model score 0.5 or higher which usually indicates the correct fold, with 48 having pTM lower than 0.5. Full scores.
Image of 466 predicted proteins colored using standard AlphaFold pLDDT colors.
Explain disk space requirements, and speed advantages of using a fast NVMe drive.
The fasta input of all the sequences should have short names as header lines because they are used as file names for MSAs and structure predictions. The command script below gives an example of reducing the Mycoplasma genome sequences fasta header lines to have short names.
I added an option to colabfold_search to create the sequence alignments for each monomer that can be used to make paired alignments for dimer predictions. This --pre_pairing option is in my fork of the Colabfold github project branch name pre_pairing.
Normal colabfold_search on a protein monomer produces a deep sequence alignment filtered down often 100-fold since prediction programs don't work with full 100,000 or 1,000,000 sequence alignments. The filtered alignment eliminates most of the homologs so pairing will find few species in common in filtered alignments of two proteins. The --pre_pairing outputs the unfiltered sequence alignment.
I wrote a Python script boltz_msa_pairing.py that creates paired sequence alignments from the protein monomer full alignments by pairing sequences from the same species. The script takes a directory of Boltz yaml input files for any number of dimer predictions and the directory containing the monomer sequence alignments, and makes new Boltz .csv paired alignment files for each dimer prediction.
I think so. The GPU mmseqs requires CUDA but the CPU mmseqs with k-mer prefiltering is almost as fast. The main slowdown would be Boltz runs slower on Mac (Nvidia 4090 about 5x faster than Mac Studio M2 Ultra). In trying it mmseqs crashed when creating the MSAs using k-mer prefiltering on the env database on a Mac with 64 GB. I added a --split-memory-limit option to colabfold_search but it did not solve this. The MSA calculation in a test with two proteins MG 7 and 11 failed with a crash in mmseqs search and is described in bug report #1037 so I did not proceed further on the Mac.
Here are example commands to carry out all the analysis shown in the Mycoplasma example above using Boltz, Colabfold search, ChimeraX, and a variety of Python scripts (scripts.zip). It includes steps to install the needed sequence databases and software. The results reported above used a Linux system with Nvidia graphics and 64 GB of memory.
mkdir boltz_screen
cd boltz_screen
mkdir databases
cd databases
# Download colabfold databases. These are about 250 GB total compressed. Can take hours to download.
curl -O https://wwwuser.gwdg.de/~compbiol/colabfold/uniref30_2302.tar.gz
curl -O https://wwwuser.gwdg.de/~compbiol/colabfold/colabfold_envdb_202108.tar.gz
tar xf uniref30_2302.tar.gz
tar xf colabfold_envdb_202108.tar.gz
# TODO: For using mmseqs GPU CUDA acceleration on Linux would want to compute the .idx* database files.
# Create mapping file from UniRef100 id to NCBI taxonomy id. Took 13 minutes.
python3 ../scripts/uniref100_taxonomy_ids.py uniref30_2302_db_seq_h uniref100_ncbi_taxonomy_ids.npz
cd ..
# Download mmseqs executable
curl -O -L https://github.com/soedinglab/MMseqs2/releases/download/18-8cc5c/mmseqs-osx-universal.tar.gz
tar xf mmseqs-osx-universal.tar.gz
# Install colabfold in Python virtual environment to use colabfold_search.
python3 -m venv colabfold
./colabfold/bin/pip install git+https://github.com/RBVI/ColabFold.git@pre_pairing
# Setup data
mkdir mycoplasma
cd mycoplasma
curl -O https://www.rbvi.ucsf.edu/chimerax/data/local-msa-sep2025/data/mycoplasmoides_genitalium.fasta
# Rewrite fasta sequence headers with short names for use in MSA file names.
python3 ../scripts/shorten_fasta_headers.py locus_tag gene < mycoplasmoides_genitalium.fasta > mgen_476.fasta
# Compute MSAs for all monomer proteins.
# The memory limit and threads option added to avoid a crash ("prefilter died") on a Mac with 64 GB.
# Was not needed on Linux with 64 GB.
mkdir msas
time ../colabfold/bin/colabfold_search mgen_476.fasta ../colabfold_databases msas --mmseqs ../mmseqs/bin/mmseqs --merge-a3m 0 --pre-pairing >& msa_476.out
# Add taxonomy info to pre-pairing MSAs to allow pairing.
python3 ../scripts/add_taxonomy_to_a3m.py ../colabfold_databases/uniref100_ncbi_taxonomy_ids.npz msas/*.pre_paired.a3m
# Create Boltz yaml input files to predict dimers.
# Use ChimeraX version newer than Sept 20, 2025.
# Enter the following commands on ChimeraX command line:
cd /path/to/mycoplasma-directory
alphafold dimers mgen_476.fasta maxLength 1200 maxSpacing 1 outputYaml boltz_dimers msaDir msas
# Make the paired MSAs needed for the dimer predictions.
python3 ../scripts/boltz_msa_pairing.py boltz_dimers msas
# Run the Boltz predictions.
# Make sure Boltz has been installed by ChimeraX using ChimeraX menu Tools / Structure Prediction / Boltz
# and press the "Install Boltz" button on the Boltz panel. The button is only there if you have not yet
# installed Boltz.
cd boltz_dimers
~/boltz22/bin/boltz predict ../boltz_dimers >& boltz.out
# Make table of confidence scores (e.g. ipTM) for dimers.
python3 ../../scripts/iptm_dimer_scores.py boltz_results_boltz_dimers/predictions > dimer_scores.txt
# To predict monomers make the Boltz yaml input files with ChimeraX command "alphafold monomers".
cd ..
mkdir boltz_monomers
# Use ChimeraX version newer than Sept 20, 2025.
# Enter the following commands on ChimeraX command line:
cd /path/to/mycoplasma-directory
alphafold monomers mgen_476.fasta maxLength 1200 outputYaml boltz_monomers msaDir msas
# Then run Boltz to predict the monomers
cd boltz_monomers
~/boltz22/bin/boltz predict ../boltz_monomers >& boltz.out
# To make a tiled image monomers start ChimeraX and use commands in ChimeraX.
cd /path/to/mycoplasma-directory/boltz_monomers
open boltz_results_boltz_monomers/predictions/*/*.cif
color bfactor palette alphafold
tile spacing 1.0
label all model
# To strip off _model_0.cif suffixes in labels open the following Python in ChimeraX
open ../../scripts/shorten_model_names.py
label all model
# To save image
save monomers.png width 3000
# To display a monomer MSA in ChimeraX as a Profile Grid you first have to convert the .a3m file to fasta.
# The .a3m files differ only in that lower case letters in the sequence indicate insertions. Deleting
# the lower case letters make it a fasta file.
cd /path/to/mycoplasma
python3 ../scripts/a3m_to_fasta.py msas/MG_007.a3m
# In ChimeraX use command
open /path/to/mycoplasma/msas/MG_007.fasta viewer grid
# To show a paired MSA as a ChimeraX profile grid you need to produce a paired fasta file.
cd /path/to/mycoplasma/
python3 ../scripts/fasta_msa_pairing.py msas/MG_007.pre_paired.a3m msas/MG_011.pre_paired.a3m > msas/MG_007_011.fasta
# Then open that fasta in ChimeraX with ChimeraX command
open /path/to/mycoplasma/msas/MG_007_011.fasta viewer grid