Structure prediction with Boltz 2 and ChimeraX
Tom Goddard
Biophysics 204B: Methods in Macromolecular Structure, 2 weeks on structure prediction and design
January 8, 2026
January 8 agenda
If the predictions don't work for you here is a zip file
ntca_predictions.zip (5 MB)
so you can look at the results.
Why Boltz instead of Alphafold 3 or others?
- Boltz looks like the best effort by and for academic researchers.
- Also academic RosettaFold from David Baker lab at Univ Washington, more oriented to testing new methods than production code.
- Other programs are very commercial, mostly open source, but little community development:
- AlphaFold 3 (Google)
- Chai (SF startup Chai Discovery)
- Paddle Helix (Baidu, the Chinese Google)
- Protenix (ByteDance owner of TikTok)
- ESMFold (Meta)
- SimpleFold (Apple)
- OpenFold (non-profit consortium of dozens of big and small pharma companies)
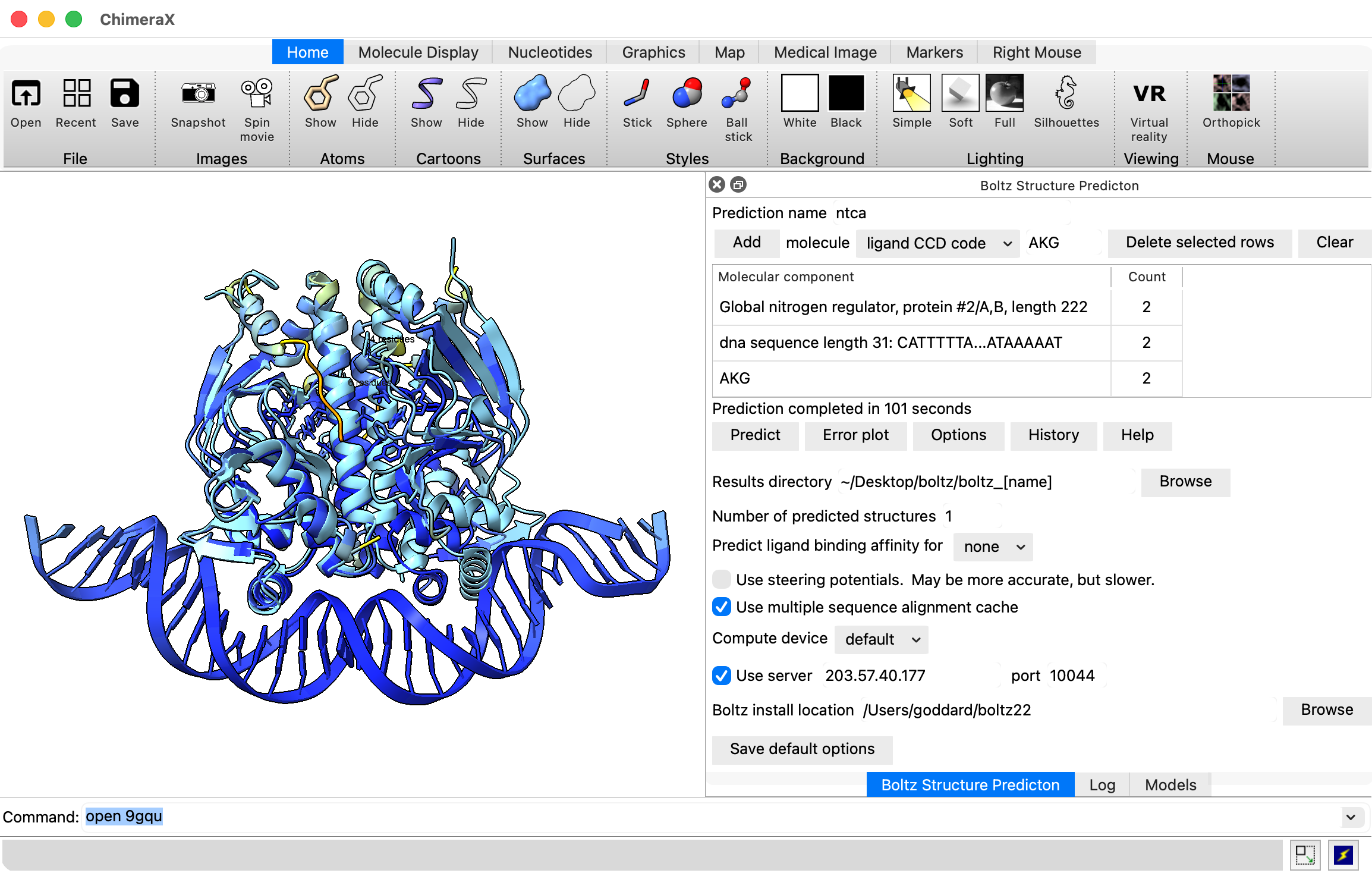
Predict NTCA monomer on MacAir laptops, look at pLDDT, PAE.
- NTCA binds to a DNA promoter when nitrogen levels are low in cyanobacteria.
Binding is enhanced 5-fold by small moleclue 2-oxoglutarate binding to NTCA.
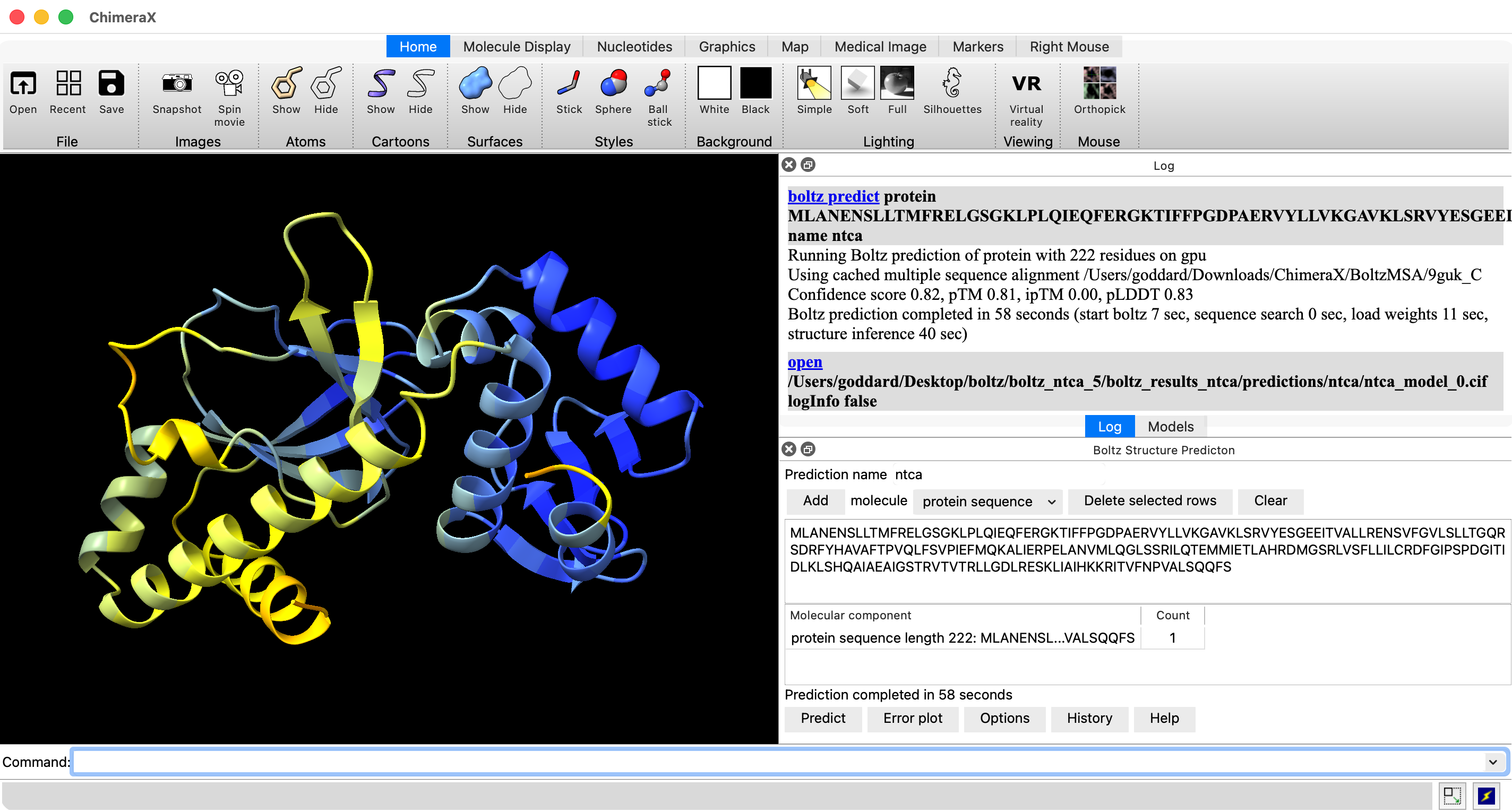
- Start ChimeraX (use daily build from 2026 for later Boltz server usage).
- Show Boltz user interface, menu Tools / Structure Prediction / Boltz.
- If Boltz not yet installed click "Install Boltz" button. Takes several minutes.
- Paste in NTCA sequence from UniProt database NTCA_SYNE7 and press Add button: MLANENSLLTMFRELGSGKLPLQIEQFERGKTIFFPGDPAERVYLLVKGAVKLSRVYESGEEITVALLRENSVFGVLSLLTGQRSDRFYHAVAFTPVQLFSVPIEFMQKALIERPELANVMLQGLSSRILQTEMMIETLAHRDMGSRLVSFLLILCRDFGIPSPDGITIDLKLSHQAIAEAIGSTRVTVTRLLGDLRESKLIAIHKKRITVFNPVALSQQFS
- Set prediction name to ntca.
- Press predict. Takes 2-4 minutes on MacAir laptop.
How did ChimeraX run Boltz?
- ChimeraX installed Boltz in ~/boltz22 (basically it makes a Python virtual environment "python -m venv ~/boltz22" then "~/boltz22/bin/pip install https://github.com/RBVI/boltz/archive/chimerax_boltz22.zip")
- Boltz neural network parameter get downloaded to ~/.boltz
- Your prediction was run in a directory ~/Desktop/boltz_ntca by running the ~/boltz22 executable as a separate process.
- ChimeraX created a Boltz input text file ~/Desktop/boltz_ntca/ntca.yaml
- Then it ran the boltz executable using the command in ~/Desktop/boltz_ntca/command
What is in the .yaml input file?
version: 1
sequences:
- protein:
id: [A]
sequence: MLANENSLLTMFRELGSGKLPLQIEQFERGKTIFFPGDPAERVYLLVKGAVKLSRVYESGEEITVALLRENSVFGVLSLLTGQRSDRFYHAVAFTPVQLFSVPIEFMQKALIERPELANVMLQGLSSRILQTEMMIETLAHRDMGSRLVSFLLILCRDFGIPSPDGITIDLKLSHQAIAEAIGSTRVTVTRLLGDLRESKLIAIHKKRITVFNPVALSQQFS
Many other things can go in (DNA, RNA, Ligands, restraints, templates, MSAs, ...)
as shown in the Boltz github file
docs/prediction.md
What output files were produced?
- Results in subdirectory ~/Desktop/boltz_ntca/boltz_results_ntca
- Multiple sequence alignment (MSA) in "msa" subdirectory (unless ChimeraX cached MSA used, then in ~/Downloads/ChimeraX/BoltzMSA).
- Structure and confidence scores in "predictions/ntca" subdirectory: structure (.cif), confidence (.json), predicted aligned error (pae), predicted distance error (pde) and predicted local distance different test (plddt) all (.npz, numeric python (numpy) array file format).
Prediction results ChimeraX shows
- pLDDT confidence coloring per residue. Average all residues 0.83 (good) on 0-1 scale.
- 100
to 90
– high accuracy expected
- 90
to 70
– backbone expected to be modeled well
- 70
to 50
– low confidence, caution
- 50
to 0
– should not be interpreted, may be disordered
- Predicted template model score pTM 0.82
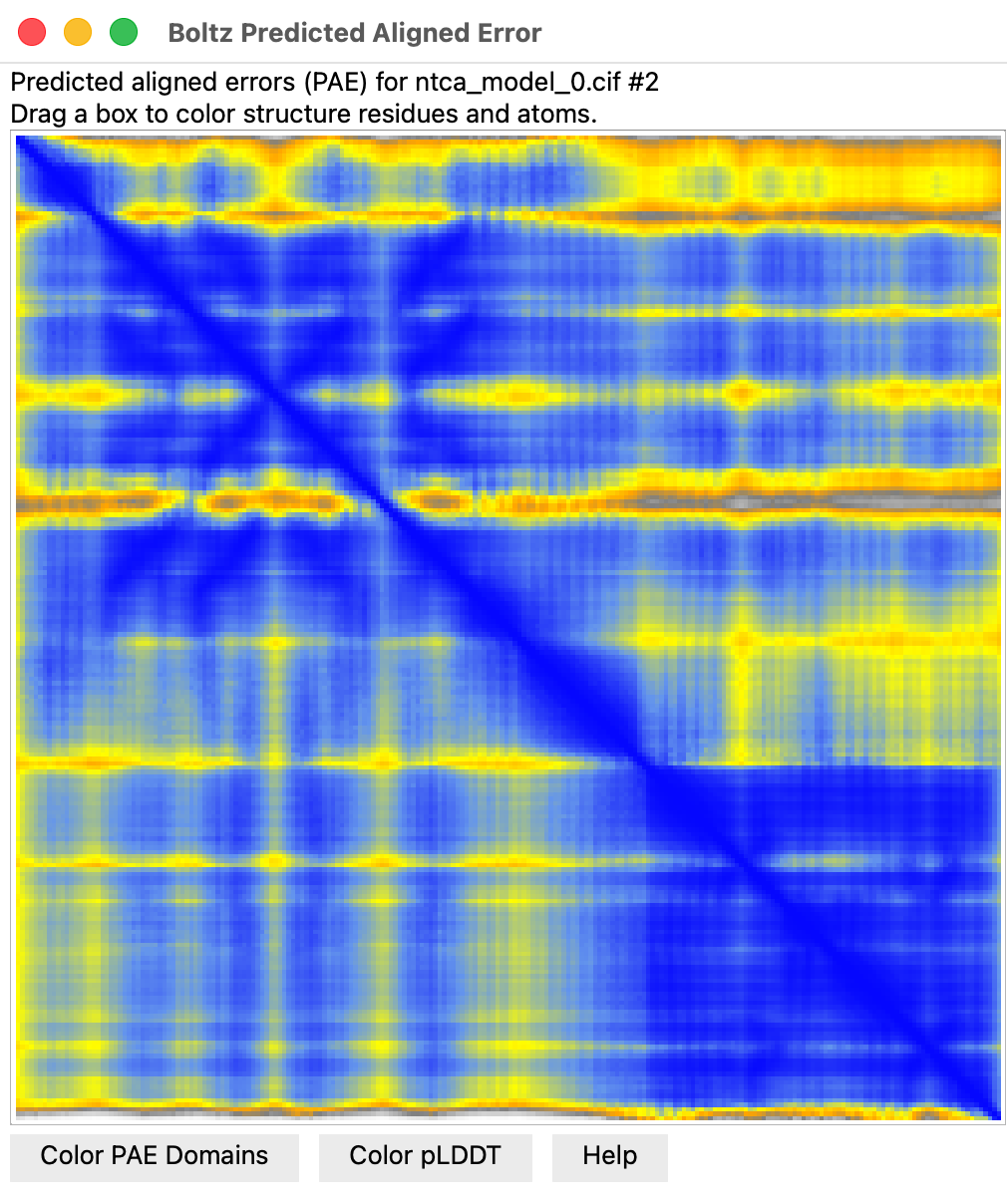
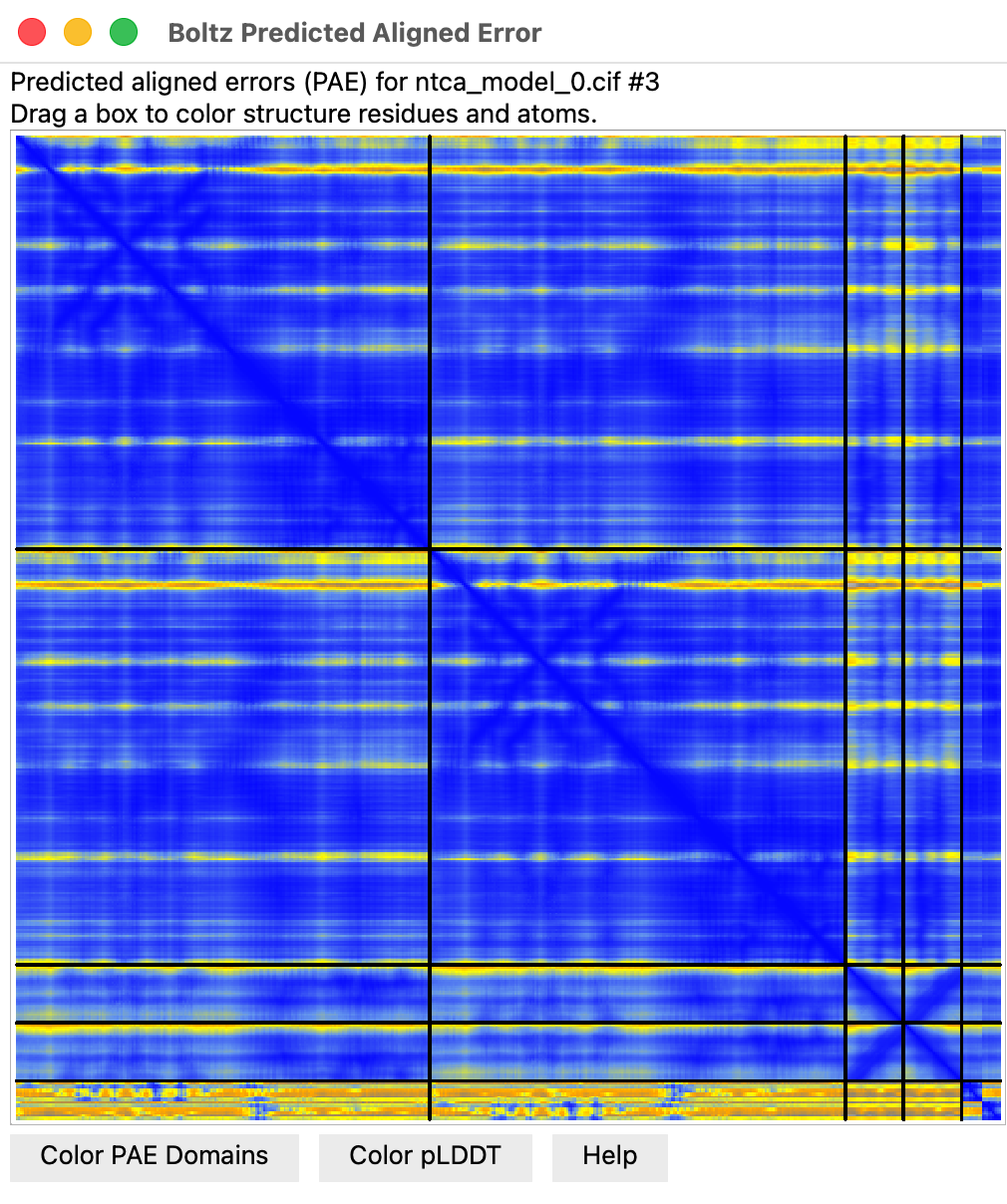
- Predicted aligned error plot (Error Plot button) score for each residue pair
linearly interpolated
Can Boltz predict NTCA correctly bound to DNA?
- Now let's try the NTCA dimer, with duplex DNA, and the 2-oxoglutarate low-nitrogen indicator.
- A 16 GB MacAir may barely be able to run this without running out of memory, might take 10 minutes.
- For bigger predictions we can use Linux Nvidia graphics cloud virtual machine.
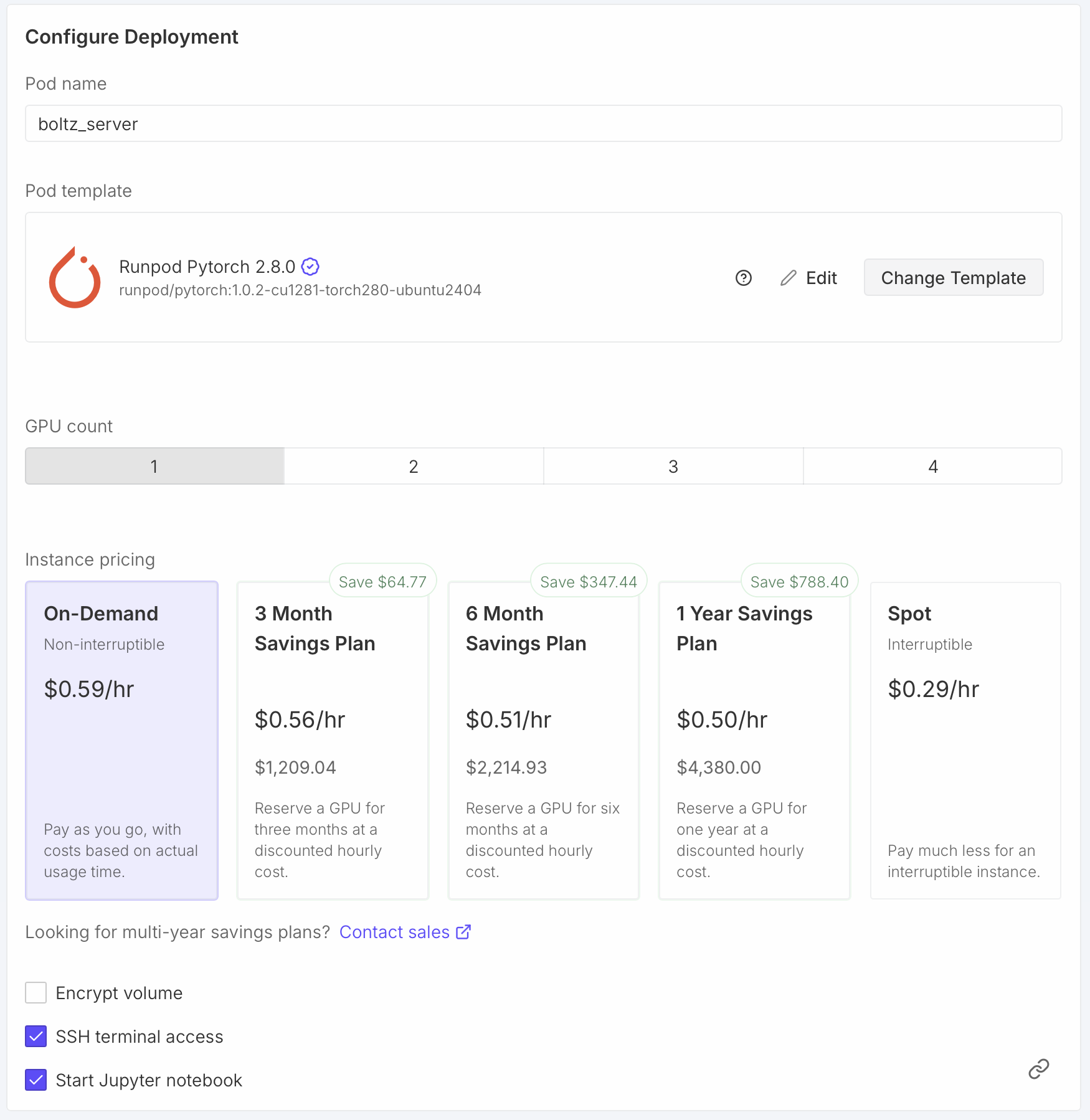
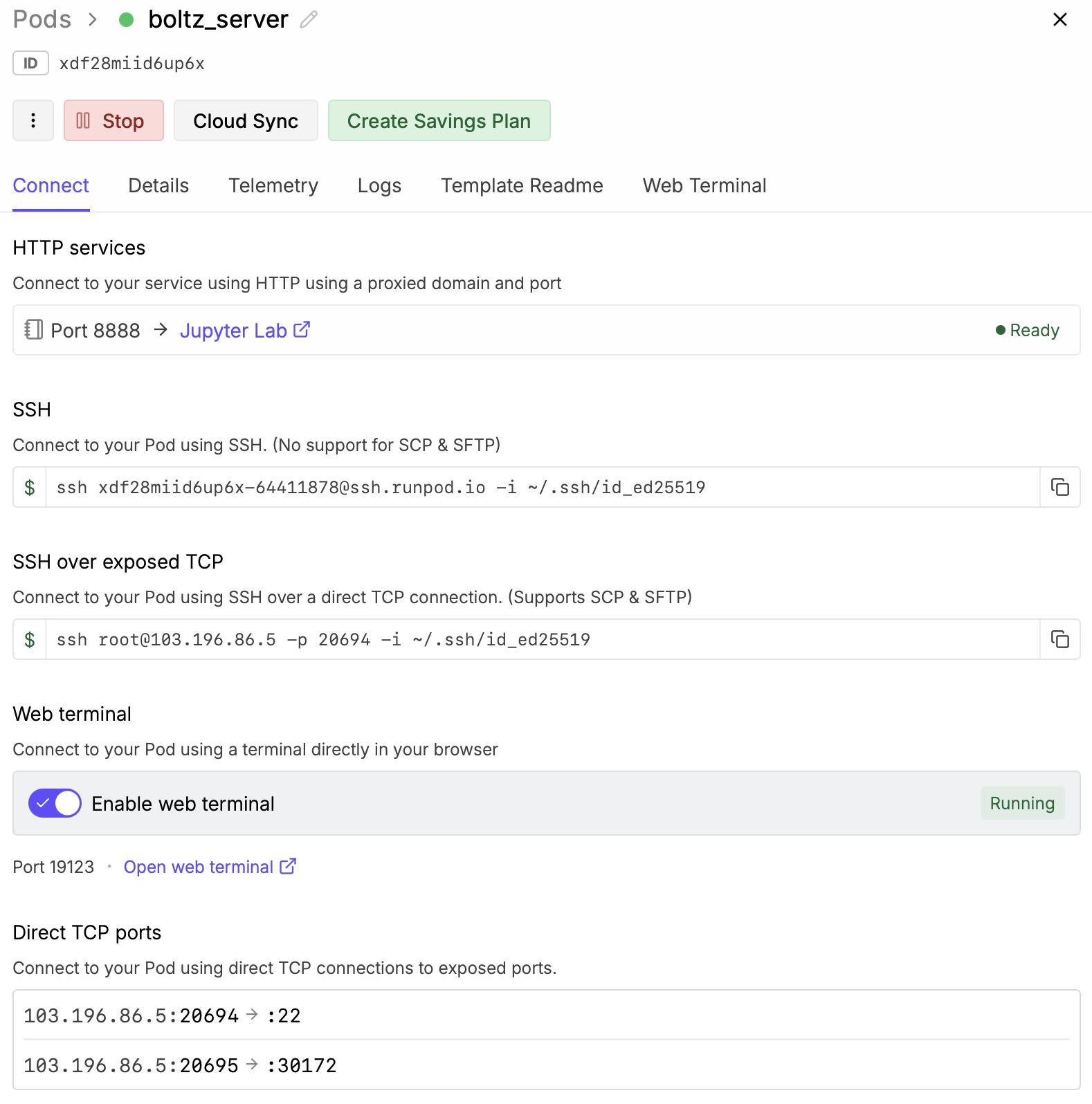
Each student spin up their own virtual machine on RunPod to do larger predictions.
- We'll use Nvidia 4090 GPUs on commercial service RunPod.io
to predict NTCA binding.
- Login to RunPod: I'll give you the login and password for our class account in class.
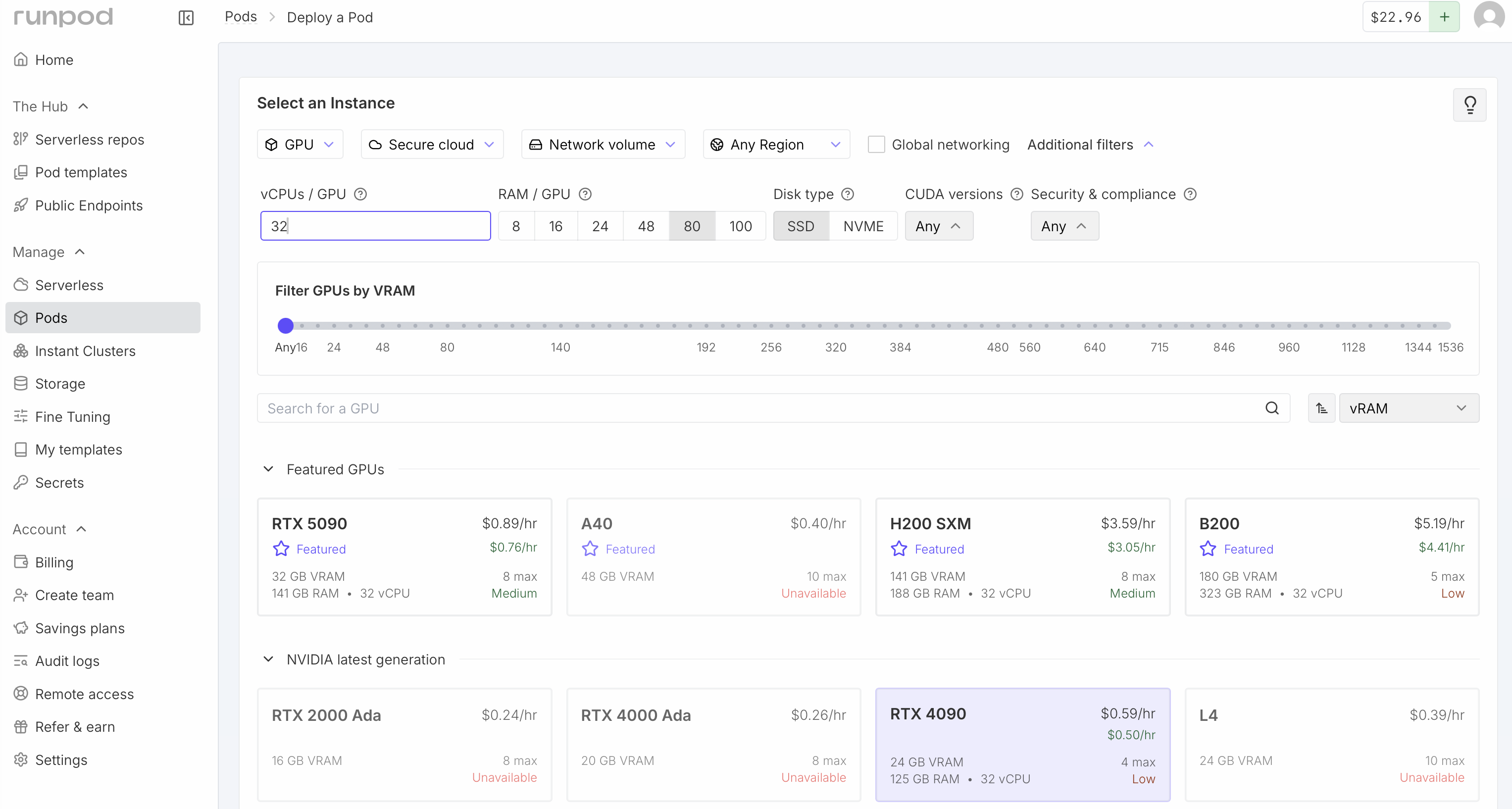
- Click Pods at the left side of the RunPod web page.
- Under the Network Volume select "Boltz and BoltzGen"
- Click additional filters near the top and request at least 8 vCPU and at least 48 GB RAM.
- Click RTX 4090
- Scroll down to Configure Deployment and set the Pod name to your name.
This will help you later see which is your VM when many are running.
- Click the "Change Template" button and choose Runpod Pytorch 2.8.0.
This will use newer possibly faster CUDA library 12.8 instead of 12.4 to run Boltz on the GPU.
- Click the "Edit" button and change Expose TCP Ports from "22" to "22,30172" and click "Set Overrides".
This will allow our Boltz server to listen on port 30172 and be reached from other computers.
|
|
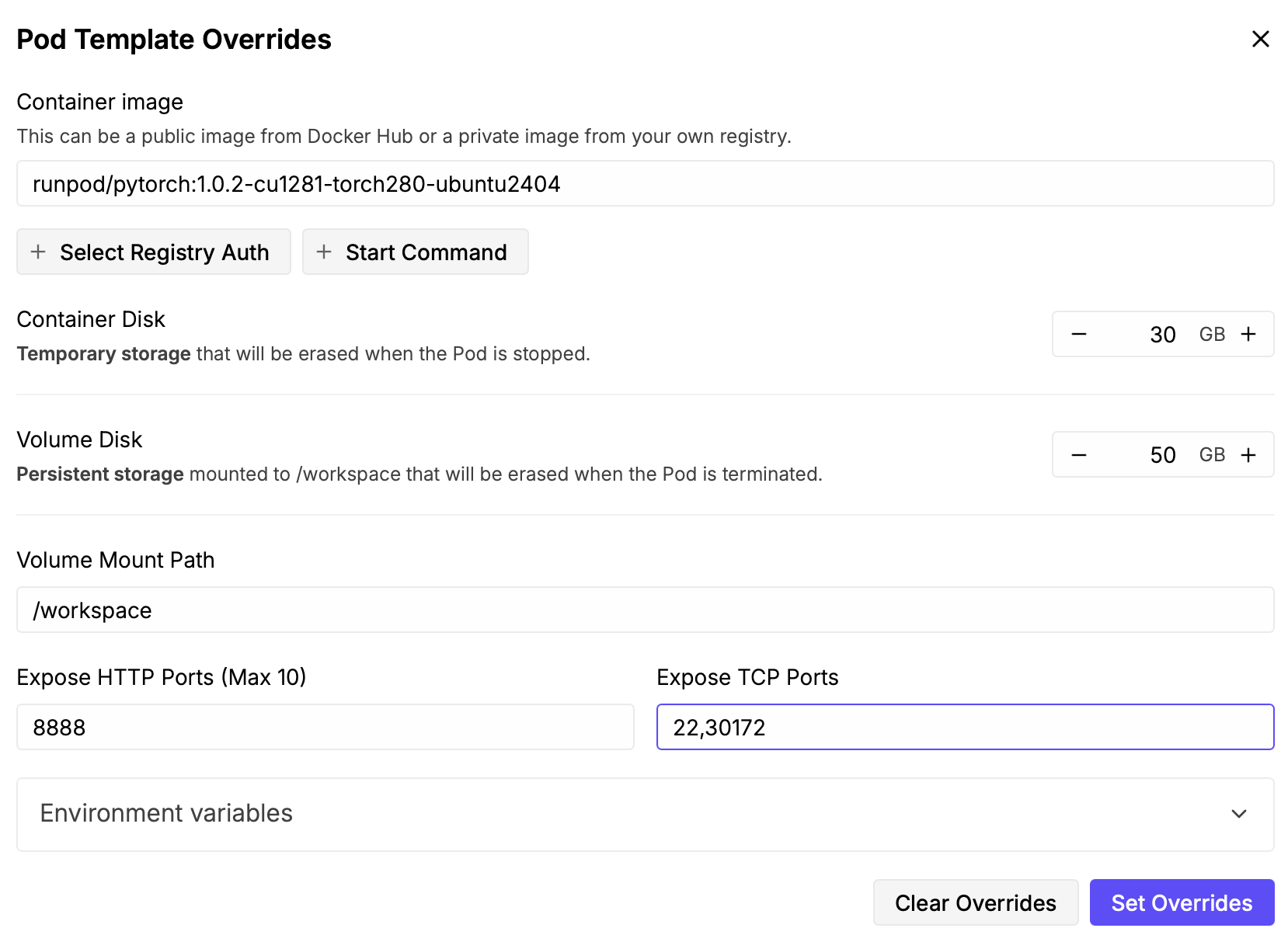
Dialog shown after pressing Edit button for Pod template
|
|
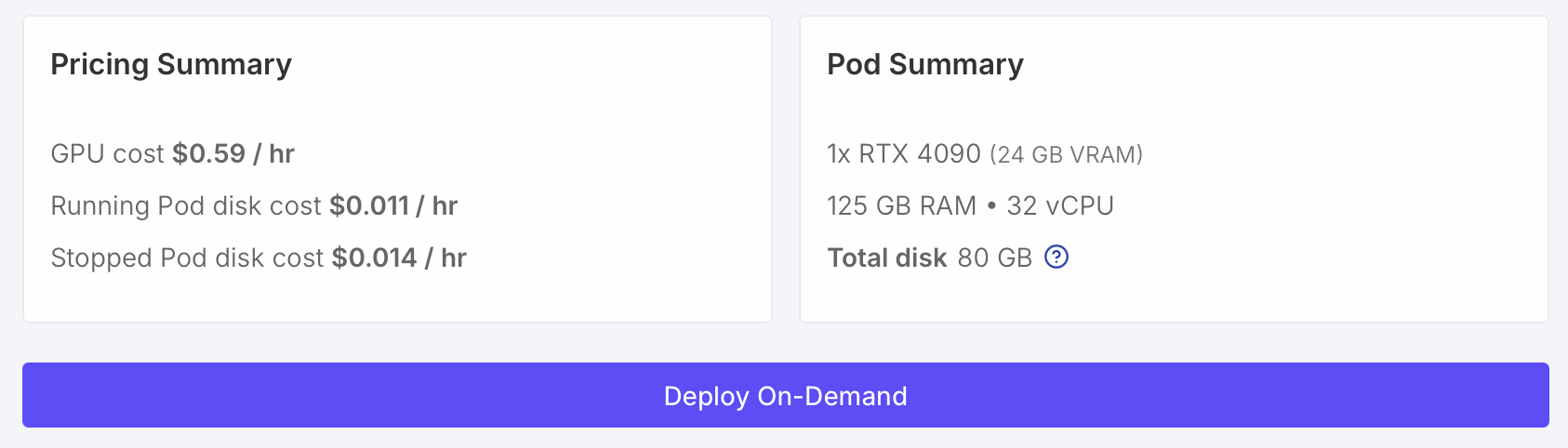
- Start the virtual machine by clicking "Deploy On-Demand". It will take about 30-60 seconds.
- Click "Enable web terminal" and then "Open web terminal" to get a shell on the virtual machine.
Run NTCA dimer with DNA duplex and 2-oxoglutarate, compare to experimental structure.
- Press the Clear button on the ChimeraX Boltz panel to start a new molecule description.
- We'll get the NTCA sequence a different way this time.
Open PDB 9gqu with ChimeraX command "open 9gqu".
- Select #2 from the Boltz molecule menu and click Add to add the dimer sequence to the prediction.
- Choose molecule "DNA sequence" paste CATTTTTATGTATCAGCTGATACATAAAAAT and press Add twice for two copies.
Palindromic DNA binds to itself (note TTTTT at 5' end pairs with AAAAA and 3' end).
- Choose "ligand CCD code" and type AKG (code for 2-oxoglutarate) and press Add twice to add 2 copies.
- Press the Options button and check "Use server host 103.196.86.5 port 20695" but put in the
host IP address and port for your virtual machine listed under Direct TCP ports in the RunPod setup
web browser page, e.g. 103.196.86.5:20695 -> 30172. This says which visible port on your VM maps to
port 30172, which is the default port used the Boltz server you started.
- Press Predict.
If you get an error right after submitting the prediction
Sometimes running a prediction reports a "RuntimeError", or in newer ChimeraX daily builds (Jan 9, 2026)
it will "Did not receive job id from server ip-address:port". This seems to be caused by
UCSF security software installed on your laptop blocking the Boltz server sending the job id back to
ChimeraX. You may be able to work around this using "ssh port forwarding". This means you tell ChimeraX
to use a Boltz server running on your laptop (localhost, port 30172) and ssh will connect that to the
actual RunPod host and port the Boltz server runs on. Here is how to setup the port forwarding.
- In the RunPod virtual machine web terminal, after you have started the Boltz server (nohup python3 ...)
find out what internet address it is using by typing command
cat ~/boltz_server_log
Boltz server listening at 172.16.112.2, port 30172
- Look at the virtual machine ssh address and port in the RunPod Details web page under "SSH over exposed TCP".
In the above example image that address and port is 103.196.86.5 and port 20694.
- Download the private ssh key file id_ed25519.txt the class is using for RunPod
(we did this step on Jan 13 in class and you don't need to repeat it). If you download the key file you
also need to set permissions on it so only you can read it, otherwise ssh won't accept it.
chmod go-w ~/Downloads/id_ed25519.txt
- On your laptop create the ssh port forwarding (also called an "ssh tunnel") using the Boltz server address
and the virtual machine ssh address and port.
ssh -Nf -L 30172:172.16.112.2:30172 root@103.196.86.5 -p 20694 -i ~/Downloads/id_ed25519.txt
- In ChimeraX in the Boltz Options panel enable "Use server localhost port 30172". You literally type
in localhost which is a standard domain name for the computer you are currently on (equivalent to 127.0.0.1).
- ChimeraX predictions will get forwarded by the ssh tunnel to the RunPod machine.
- Hopefully UCSF security software will not interfere with ssh port forwarding. Sorry this is so convoluted.
UCSF computer security often makes work difficult.
If you get a CUDA error after running the prediction
Sometimes running a prediction gives a long error traceback that mentions a CUDA problem.
Looking at the dimer prediction
- Took about 2 minutes.
- Hide experimental dimer and note the pLDDT coloring of NTCA long helix is much bluer (higher confidence).
- ChimeraX Log shows pTM 0.97 much higher than monomer, average pLDDT 0.93 higher.
- Can align the NTCA monomer prediction with ChimeraX command "matchmaker #1 to #3"
- Note that the experimental dimer has helices in the DNA groove spaced much farther apart than the prediction. Is it right?
- Compare to experimental dimer bound to DNA, PDB 9gui.
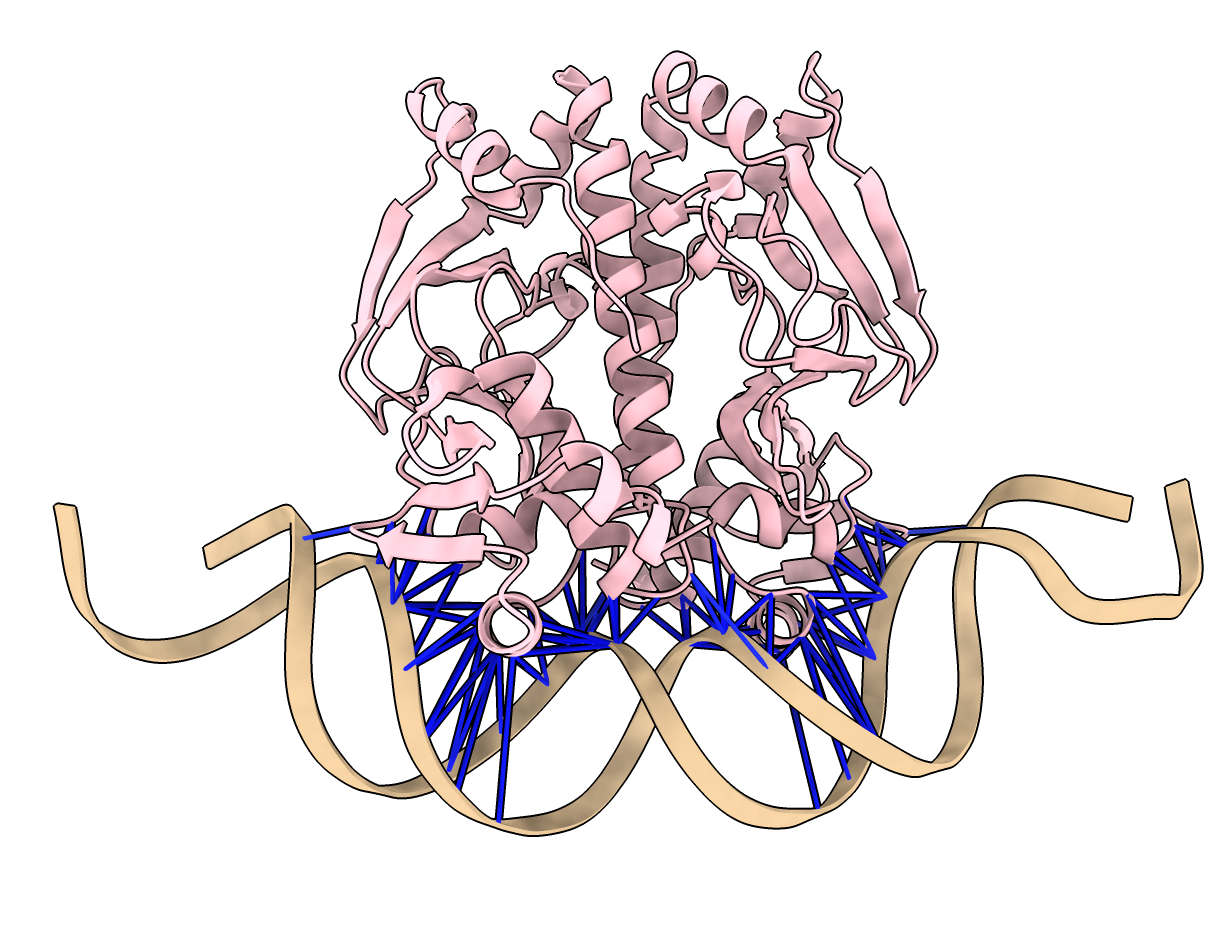
Predicted aligned error at NTCA interface with DNA
- Can show colored lines between NTCA protein residues and DNA residues colored by predicted
aligned error to see if Boltz is confident of the binding.
- Use ChimeraX alphafold contacts command.
alphafold contacts #3/A,B to #3/C,D distance 5 maxpae 100
- All blue lines indicate high confidence PAE values.