NIAID progress report for group meeting July 14 (last report) - August 24, 2022.
Tom Goddard
August 24, 2022
NIH 3D Pipeline
- Eric can describe progress.
- Gave Michal Stolarczyk code to report map voxel size and grid size.
- Advised Michal on how to get molecular weight for EMDB maps.
DICOM
- Zach can describe progress.
AlphaFold Database version 3
- DeepMind and EBI released version 3 (also called release 4) of the AlphaFold database a month ago, July 28, 2022.
- Database now has 214 million predicted structures, had just 1 million in version 2.
- Version 3 took some shortcuts. Predict just one model instead of 5. Maximum sequence length 1280 instead of 2700.

Fast Searching of 200 Million AlphaFold Models
- ChimeraX BLAST search of AlphaFold DB took about 5 seconds with version 2. Test took 20 minutes with version 3.
- Tests showed BLAST with 4 threads is 4x faster, so Zach changed to use 4 threads.
- ChimeraX AlphaFold BLAST now takes about 5 minutes for length 268 sequence.
- BLAST of length 230 sequence on Mac with fast SSD drive took 10.5 minutes with 1 thread.
- ChimeraX BLAT search to find best matching AlphaFold DB model could match several chains per second in version 2, but test took 1 hour per sequence in version 3.
- Tried mmseqs2 for fast search, most used tool for large fast sequence searches.
- Needs several hundred gigabytes of memory.
- Could not find any fast search that used modest memory (< 8 Gbytes).
- Wrote k-mer search Python code that can search 214 million sequences in 2.0 seconds for a length 230 sequence on plato (beegfs file system), or 0.2 seconds on Mac M1 laptop.
- k-mer search only finds high-identity matches > 50% identity for long sequences (1000 aa), 70% identity for medium length (200 aa), and > 90% identity for short sequences (60 aa).
- Plan to replace ChimeraX BLAT AlphaFold search with k-mer search.
DeepFoldRNA
De Novo RNA Tertiary Structure Prediction at Atomic Resolution Using Geometric Potentials from Deep Learning
Robin Pearce, Gilbert S. Omenna, Yang Zhang
Preprint at bioRxiv from May 15, 2022.
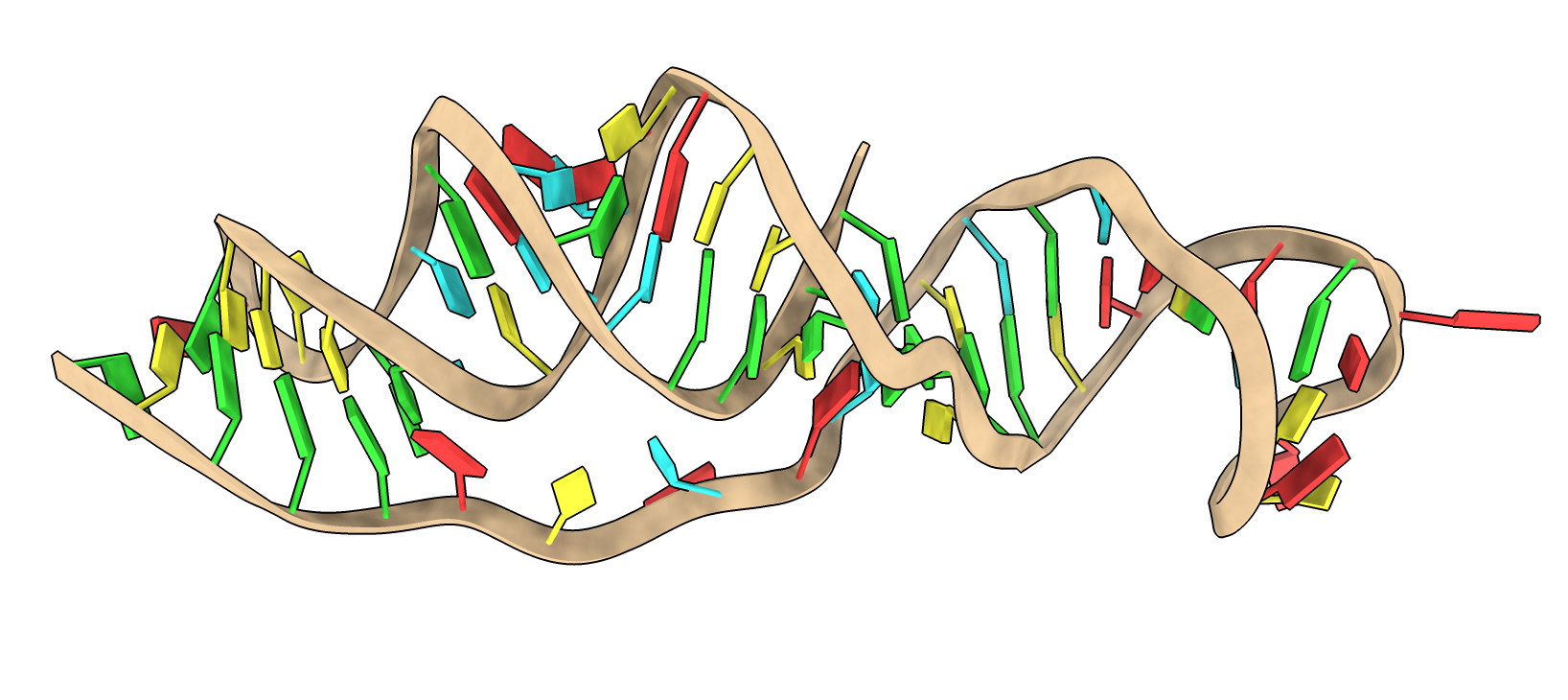
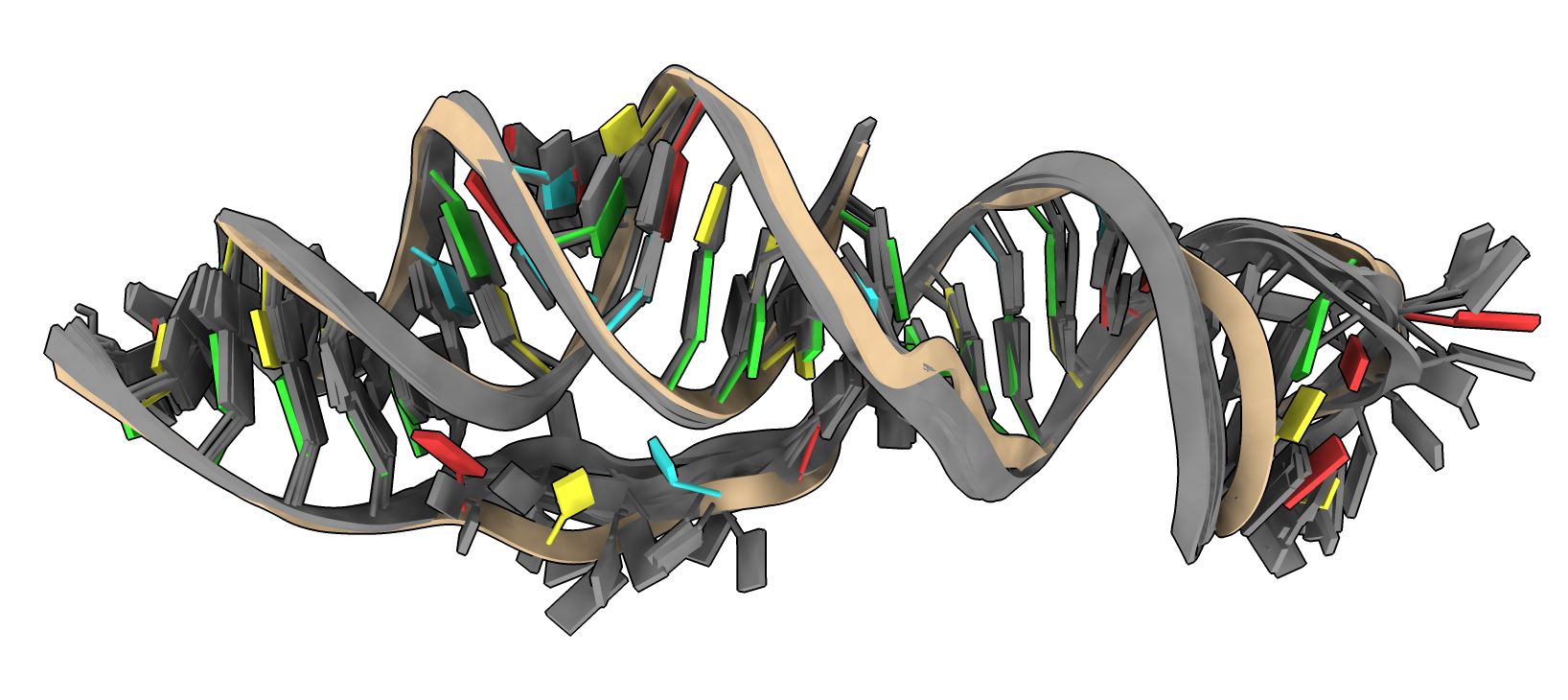
| 
|
| X-ray 7MLX
| DeepFoldRNA 5 models in gray
|
- DeepFoldRNA predicts RNA structures using similar techniques to AlphaFold.
- Predictions are typically 2-3 Angstroms from experimental RNA structures.
- Expert human theoretical modeling has only produced 10-20 Angstrom structures in the past.
- I ran DeepFoldRNA on sars-cov-2 frameshift element (65 nucleotides), agrees well with X-ray structure PDB 7mlx
- I ran on web server that took 5 days and email to author to get a result.
- We could provide a DeepFoldRNA server. Uses GPU.
- Runs take minutes according to preprint.
ESMFold
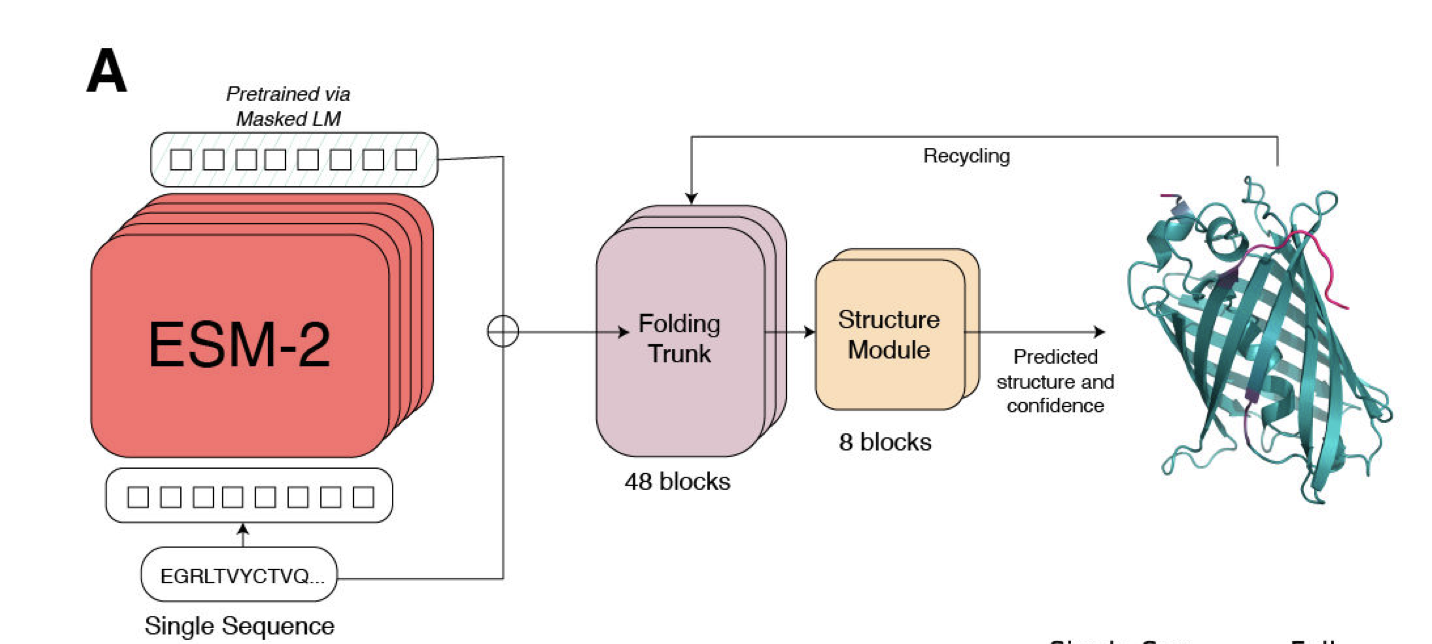
Machine learning protein structure prediction without using a deep sequence alignment.
Language models of protein sequences at the scale of evolution enable accurate structure prediction
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Allan dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Sal Candido, Alexander Rives
Preprint at bioRxiv, July 21 2022.
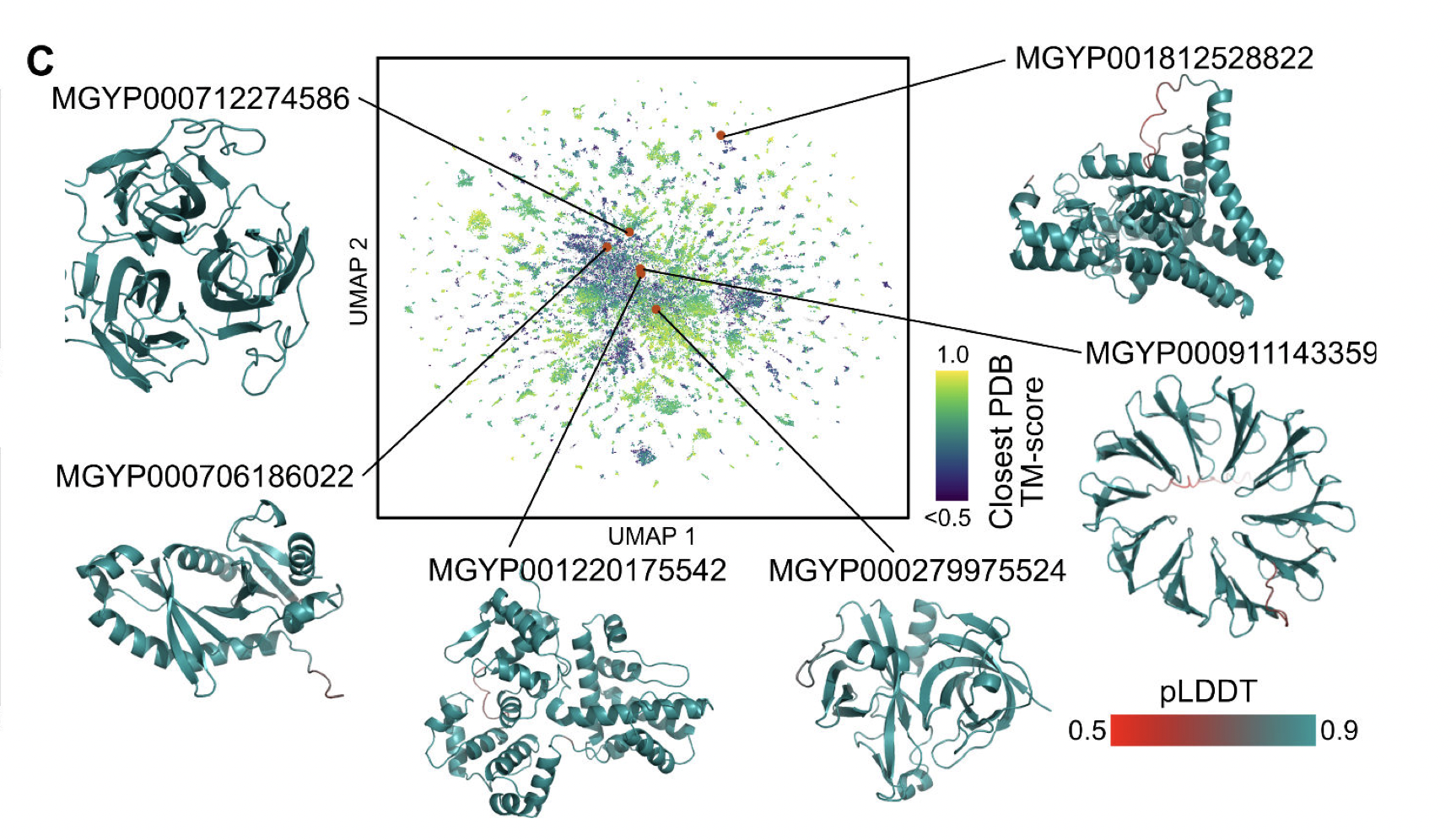
|
| UMAP of high confidence predictions from 1 million sequences
|
- Network trained by masking out 15 percent of amino acids from UniRef sequences and making the network predict the missing amino acids.
- It learns about secondary structure, contact maps, binding sites in order to predict the missing amino acids.
- This technology is used for computer interpretation of human langauge (e.g. English) where the input is a sentence.
- The high dimensional representation used by the network is then input to a network that predicts the structure.
- Results are as good as AlphaFold for about half of CASP14 sequences.
- It reliably predicts which sequences it can make accurate predictions for.
- Method runs in minutes. Uses GPU.
- Could offer a ChimeraX service to use this.
FoldSeek
Foldseek: fast and accurate protein structure search
Michel van Kempen, Stephanie S. Kim, Charlotte Tumescheit, Milot Mirdita, Johannes Soding, Martin Steinegger
Preprint in bioRxiv, Feb 9, 2022.
"To increase speed, a crucial idea is to describe the amino acid backbone of proteins as sequences over a structural alphabet and compare structures using sequence alignments [15]. Structural alphabets thus reduce structure comparisons to much faster sequence alignments.
For Foldseek, we developed a novel type of structural alphabet that does not describe the backbone but rather tertiary interactions. The 20 states of the 3D-interactions (3Di) alphabet describe for each residue i the geometric conformation with its spatially closest residue j."

- Foldseek finds structures with the same shape as a query structure in the PDB or AlphaFold DB.
- Helps find homologs whose sequences are too divergent to find by sequence search.
- Fast. About 10000 times faster than previous methods.
- Took 1 second for 77 amino acid protein against 1 million AlphaFold DB structures.
- Took 90 seconds to search 237 amino acid (2qhs) against AlphaFold DB version 3 finding
1000 matches down to 15 percent sequence identity.
- Could add a Foldseek tool to ChimeraX.
Native Mac M1 ChimeraX - Funded by CZI grant
- Mac M1 technology preview build put on download page July 22, 2022
- Tech preview downloaded 668 times by 312 unique IP addresses July 22 - August 24, 2022.
- Next step is to put universal build (ARM + Intel) up as tech preview.