ChimeraX cryoEM Visualization Tutorial: Bacterial ATP Synthase
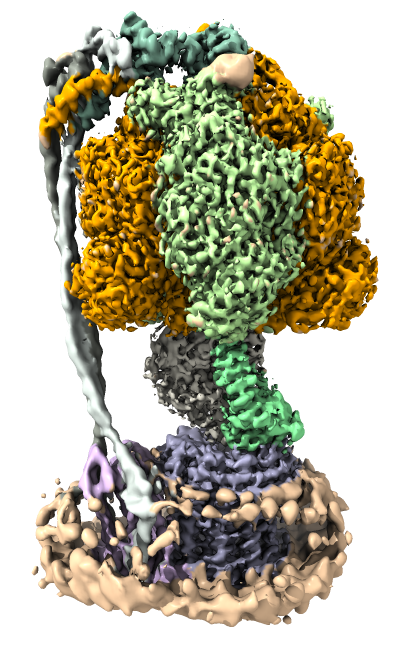
This ChimeraX tutorial will look at how to visualize atomic models and maps of
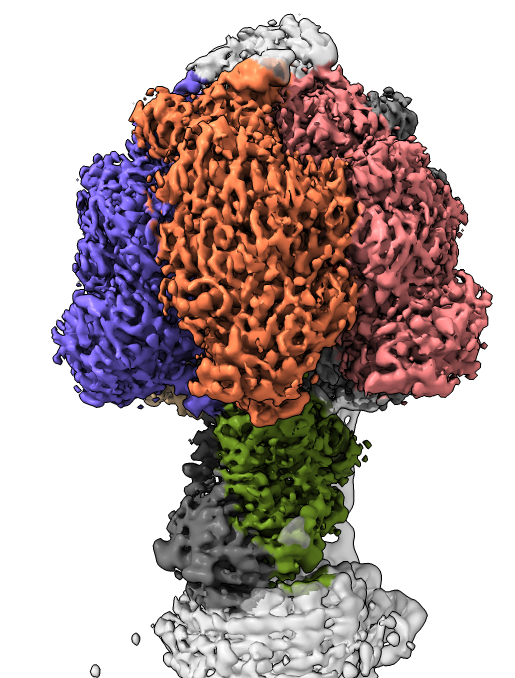
three conformations of a bacterial ATP synthase determined by cryoEM at 3.0 and 3.2 Anstrom resolution.
We will create views of the data similar to those found in the February 2019 publication.
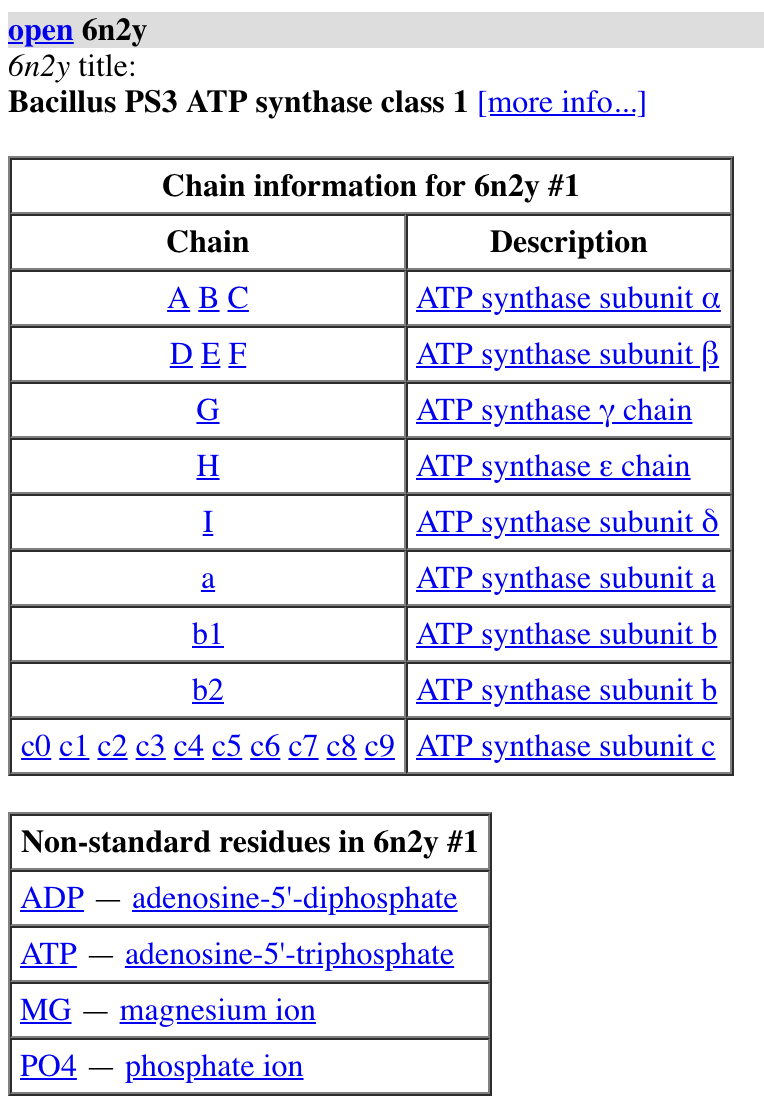
Atomic models PDB 6n2y,
6n2z,
6n30.
EMDB maps
9333,
9334,
9335.
ChimeraX can fetch these data files directly from the PDB and EMDB or you can download the files for offline use.
Topics
Basics: open, movement, styles, chains...
Morphing between 3 conformations.
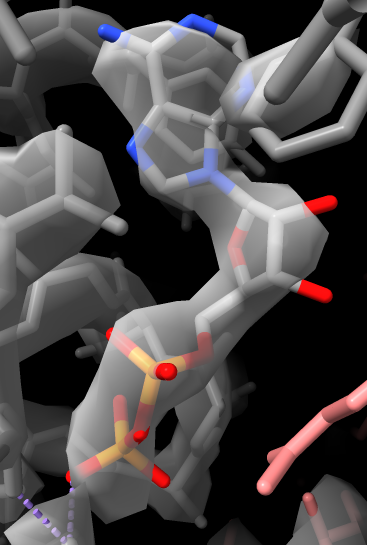
Viewing bound ADP, phosphate, ATP.
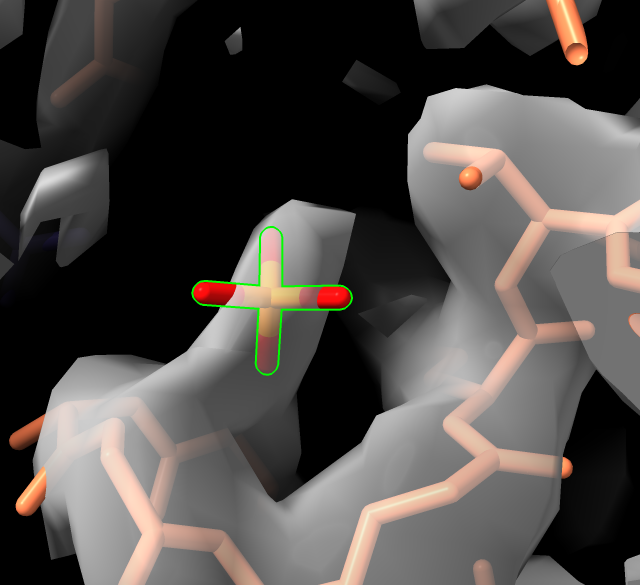
Inspecting side-chain density, zones.
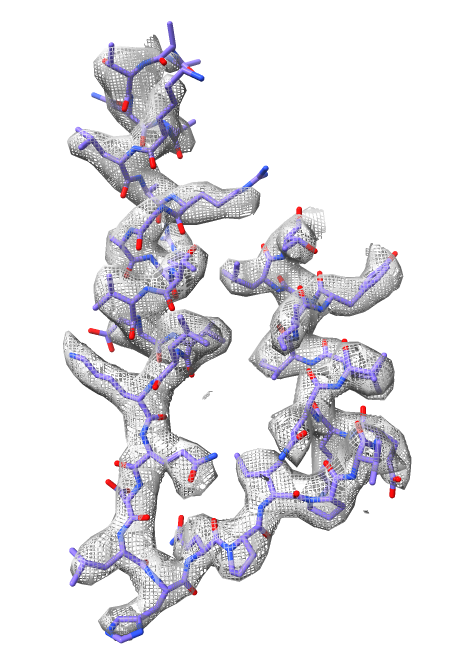
Fitting atomic model into map.
Look at proton channels.
Look at unidirectional rotation.
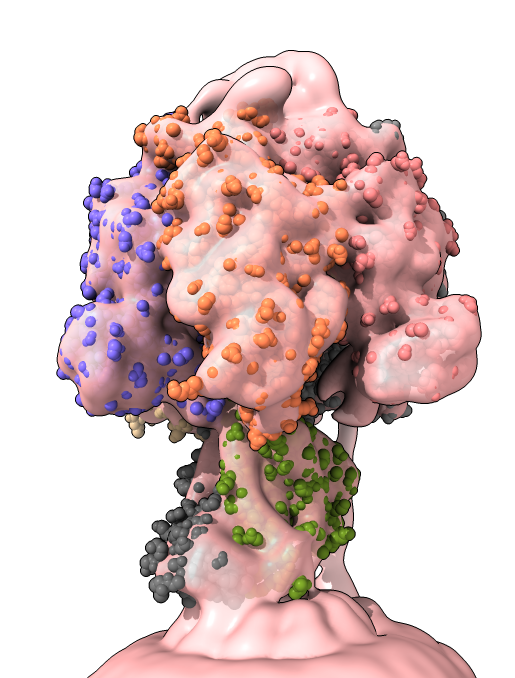
Basics: open, movement, styles, chains...
Command "open 6n2y", or toolbar Open button, or File/Open menu to load file from local drive.
Log chain and ligand tables: click letter to select atoms, click name to show sequence.
Hover mouse to show chain, residue name, number and atom name.
Movement: Mac trackpad, 2-finger rotate, 3-finger translate, 4-finger zoom (or pinch). Mouse, left rotate, right translate, scroll to zoom.
Center of rotation: on surface close up, or at center of model when zoomed out.
More info... log link, shows publication, species, experimental method and resolution.
Miscellaneous - not sure how much of this to show at the start versus later on.
Starting point for model building is often fitting existing models into map.
Fit 3.9A x-ray structure 4xd7 (F1 half of complex) into map.
Command: "open 4xd7"
Select an atom of 4xd7 by ctrl-click on it.
Use translate and rotate selection mouse modes from toolbar to place model in map.
Use menu Tools / Volume Data / Fit in Map to optimize fit of 4xd7 into 9333.
Fine detail in map causes it not to find good global fit. So fit into smoothed map.
Command "volume gaussian #4 sdev 3"
Fitting into this map has much larger radius of convergence.
Still need to get correct 6-fold symmetry alignment to match shaft subunits.
Sometimes useful to color map to match nearby atoms: "color zone #4 near #5 dist 8"
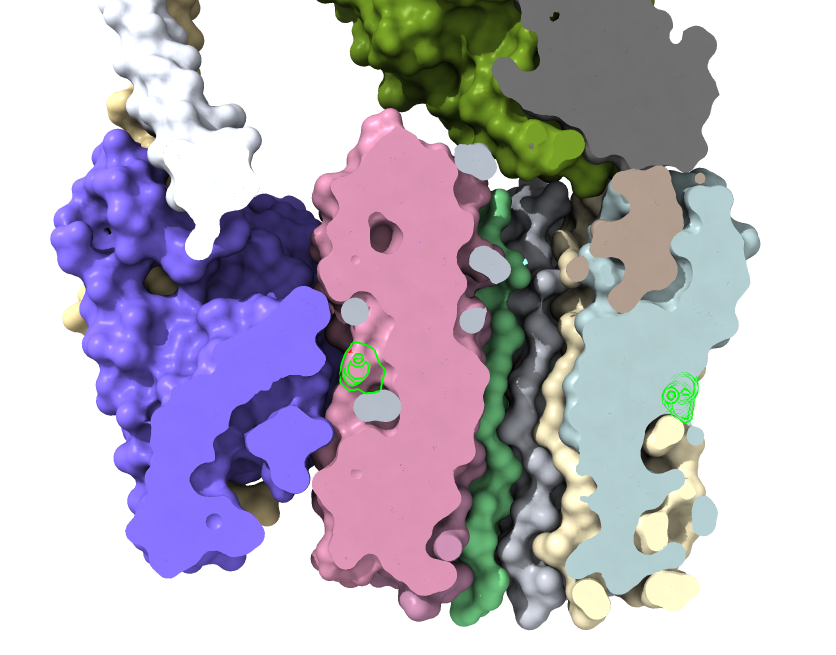
Proton intake channel.
Proton outlet channel.
Look at proton channels
Look at proton path that drives motor, figure 4 in eLife paper.
Transmembrane drum carries protons around on glutamic acid E56: "select /c*:56". Show as sphere.
Drum is in membrane and proton from lower side goes up channel between side "a" subunit, onto drum, carried around one rotation, and released into another channel to exit on other side of membrane.
Molecule toolbar, show surface.
Orient view with long helical rods on left.
Enable clip mouse mode in toolbar. Use simple lighting for faster clip plane motion.
Clip to see channel from below to E56.
Hold alt-key to clip far side to show channel releasing proton above.
After positioning clip plane turn on full lighting for darker channel cavity.
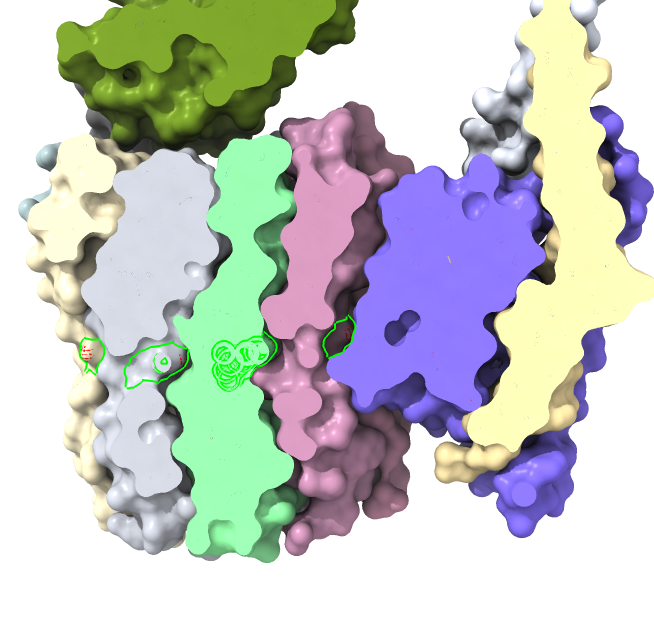
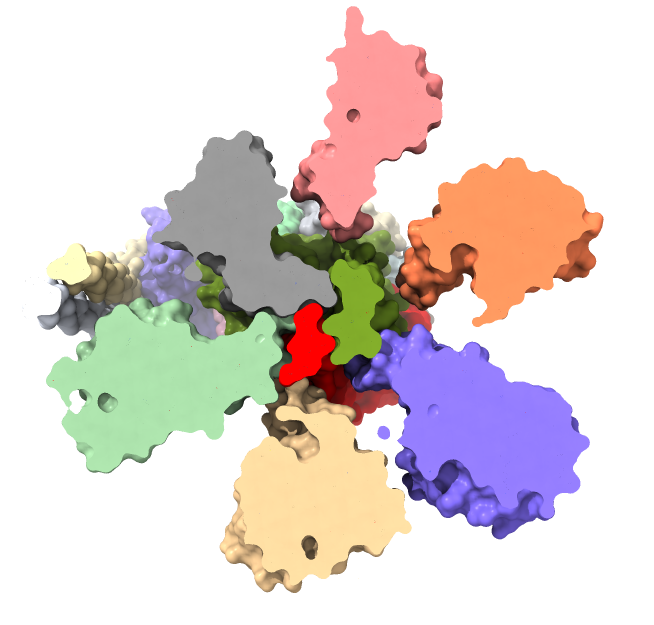
Look at unidirectional rotation
Figure 3 of eLife paper suggests mechanism why motor turns in one direction that produces ATP and is blocked in reverse direction.
Turn off clipping, command: "clip off"
Orient structure axis perpendicular to screen with F1 hexamer at front.
Make motor shaft epsilon subunit (chain H) more visible: "color /H red surface"
Clip with clip mouse mode.
Red epsilon subunit blocked from clockwise rotation by collision with beta subunit (gray color) of hexamer.