Tom Goddard
Stanford-SLAC cryoEM Center workshop
September 8, 2021
We show how to use the AlphaFold protein structure prediction to start building an atomic model in a cryoEM map using ChimeraX. We look at two examples, a possible lipid metabolism membrane protein called TACAN, and an omega-3 fatty acid transporter, both recently solved by cryoEM. We will use the AlphaFold database to fit an initial atomic model to the TACAN map, and we show how to run AlphaFold to make an initial model for the transporter example.
Video demonstrations of the steps described here are available on YouTube AlphaFold database and run AlphaFold. The written instructions below are brief and will be difficult for a new ChimeraX user to carry out, while the video shows all steps.
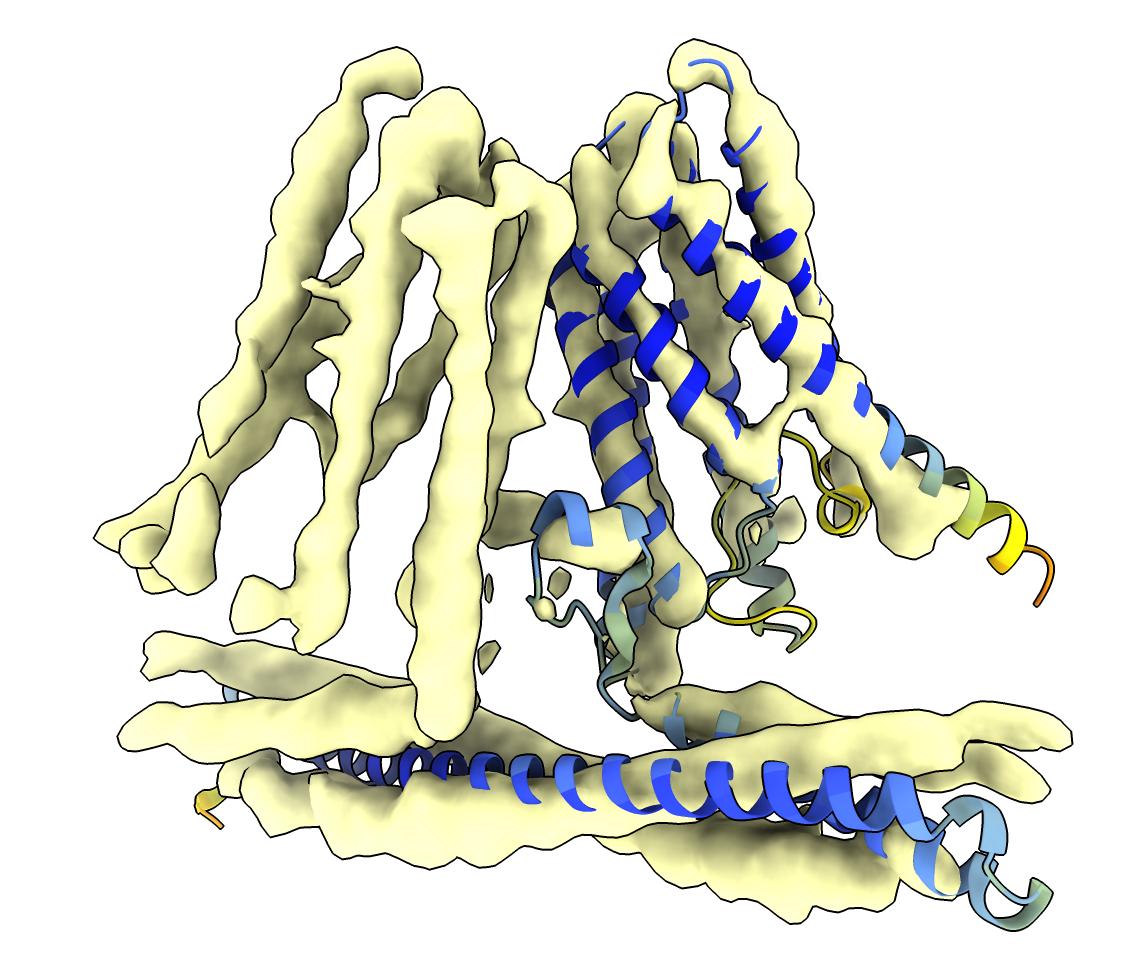
We try to find an initial atomic model for the human TACAN dimer structure seen in EMDB map 30495 using the AlphaFold database at the EBI and ChimeraX. This map and an atomic model were published last month.
Cryo-EM structures of human TMEM120A and TMEM120B. Ke M, Yu Y, Zhao C, Lai S, Su Q, Yuan W, Yang L, Deng D, Wu K, Zeng W, Geng J, Wu J, Yan Z. Cell Discov. 2021 Aug 31;7(1):77. doi: 10.1038/s41421-021-00319-5. PMID: 34465718.
A BLAST search of the PDB turns up no homolog structures prior to this structure being solved using a 20% identity or 1e-3 E-value cutoff. The structure was thought to be a mechano-sensitive ion channel involved in pain sensation but functional studies of in the above publication contradict that and they propose it is a lipid metabolism enzyme.
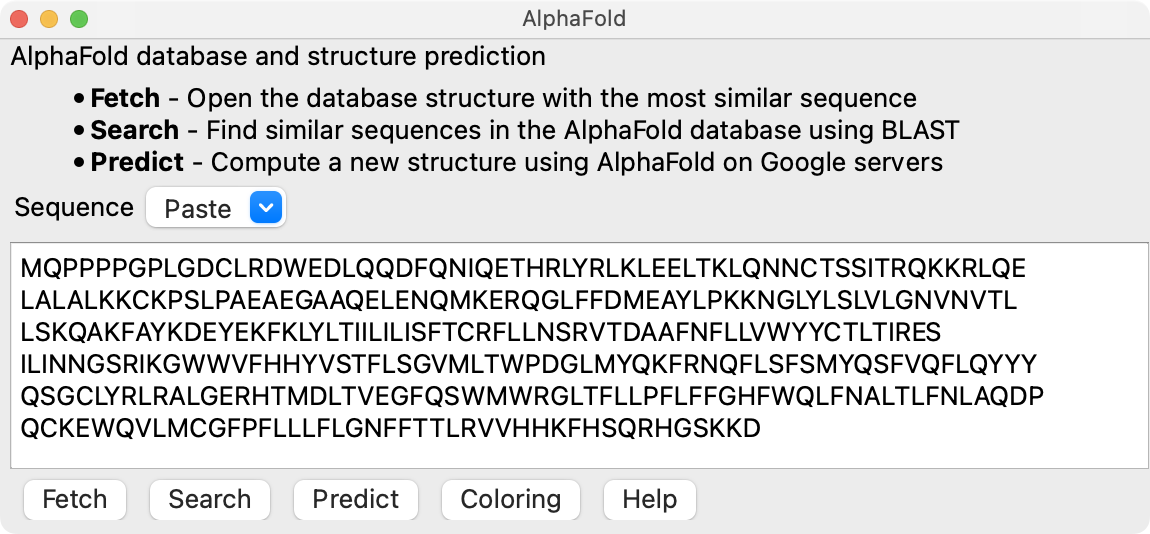
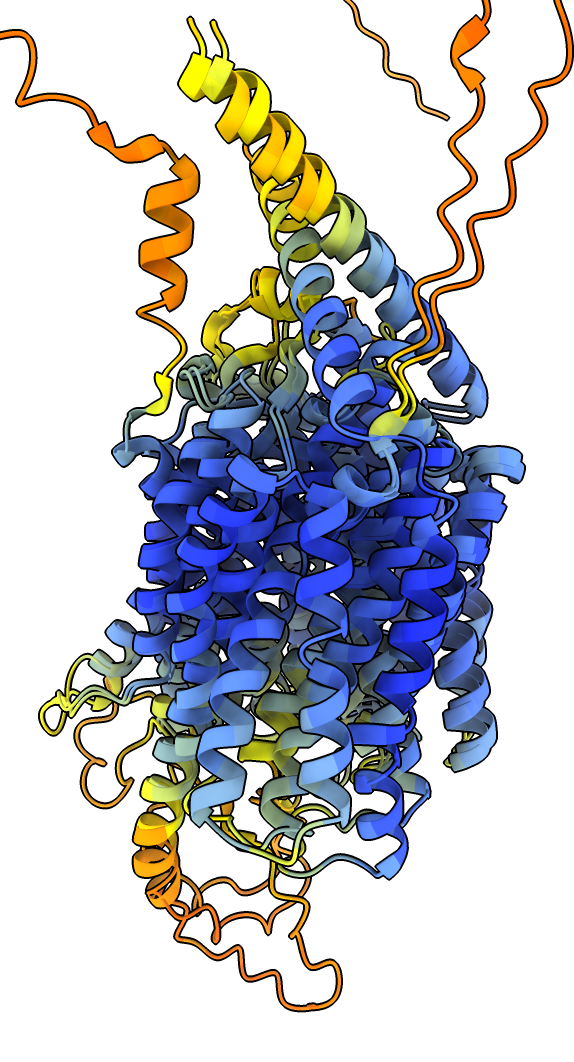
The protein is from human, uniprot sequence TACAN_HUMAN. The AlphaFold database contains precomputed structures for all genes in 21 organisms including human.
Fetch the AlphaFold database structure using ChimeraX (September 2021 version or newer) menu entry
Tools / Structure Prediction / AlphaFold
by pasting in the sequence from UniProt and pressing the Fetch button.
AlphaFold structure coloring is blue for confident regions, yellow and red less confident.
Using the Move Model mouse mode chosen from the Right Mouse toolbar, selecting the structure with the mouse with ctrl-click on the ribbon, and dragging it with the right mouse button (on Mac touchpad hold down Option key). Drag to translate structure and hold shift key to rotate it. After a rough alignment is done by hand, use ChimeraX menu
Tools / Volume Data / Fit in Map
to locally optimize the position in the map by pressing the Fit button.
|
|
|
|
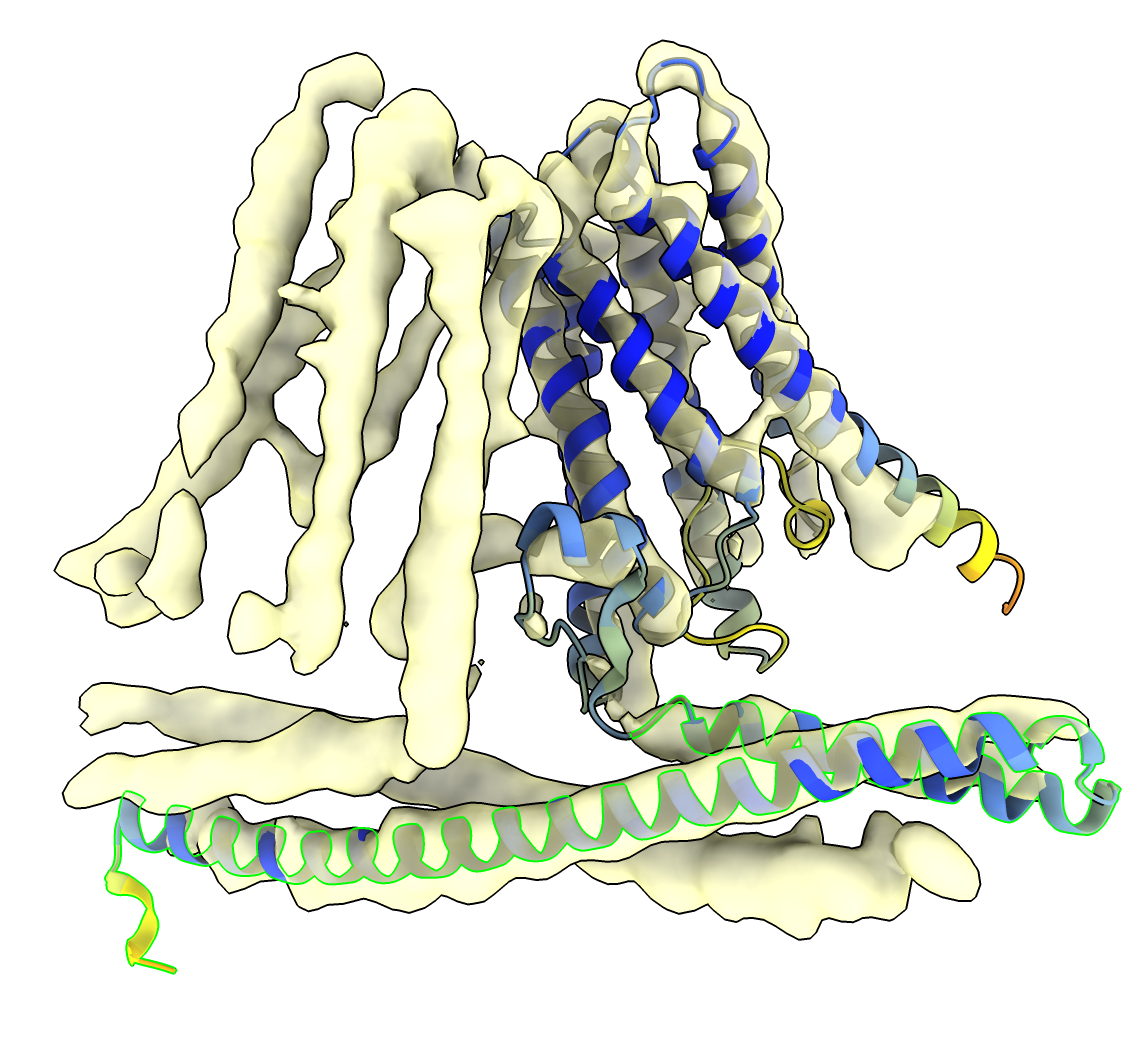
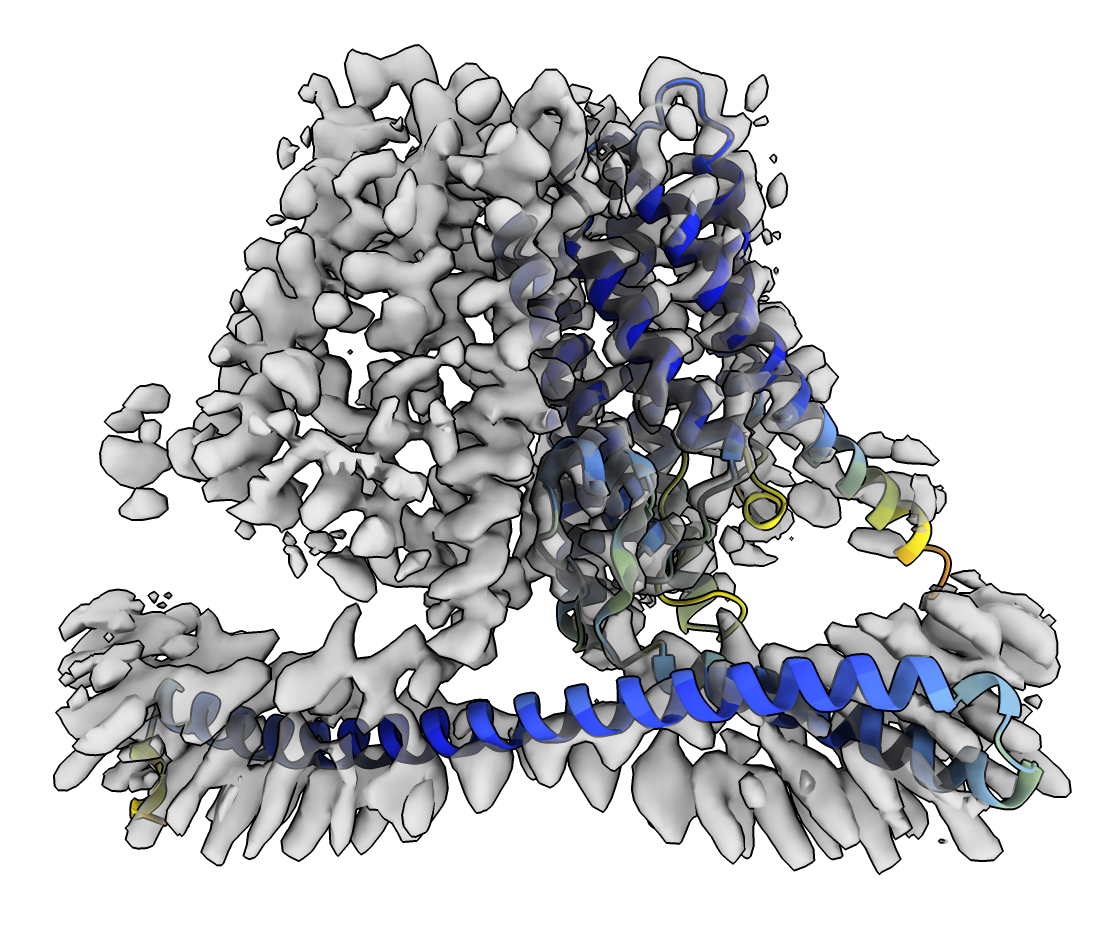
To more easily see the fit of alpha helices reduce map resolution to about 8 Angstroms with Gaussian smoothing, ChimeraX command
volume gaussian #1 sdev 2
The transmembrane helices (upper domain) fit well in the map, but the long helix at bottom is not well positioned in density.
Fit the long helix into the density using the Fit in Map panel choosing its menu entry selected atoms and selecting residues 1 to 100 with command
select :1-100
Press the Fit in Map Options button and turn off Move whole molecules so only the selected atoms are moved when the Fit button is pressed. This does puts the smaller adjoining helix in the wrong density. Manually rotate about the long axis with mouse mode Move Atoms on the Right Mouse toolbar, the press Fit again.
AlphaFold only predicts structures of single proteins, not assemblies. The position of the long helix in each monomer is controlled by its packing with the long helix in the other monomer. So it is not surprising that AlphaFold did not get this position right. It is surprising that it got the position as close as it did.
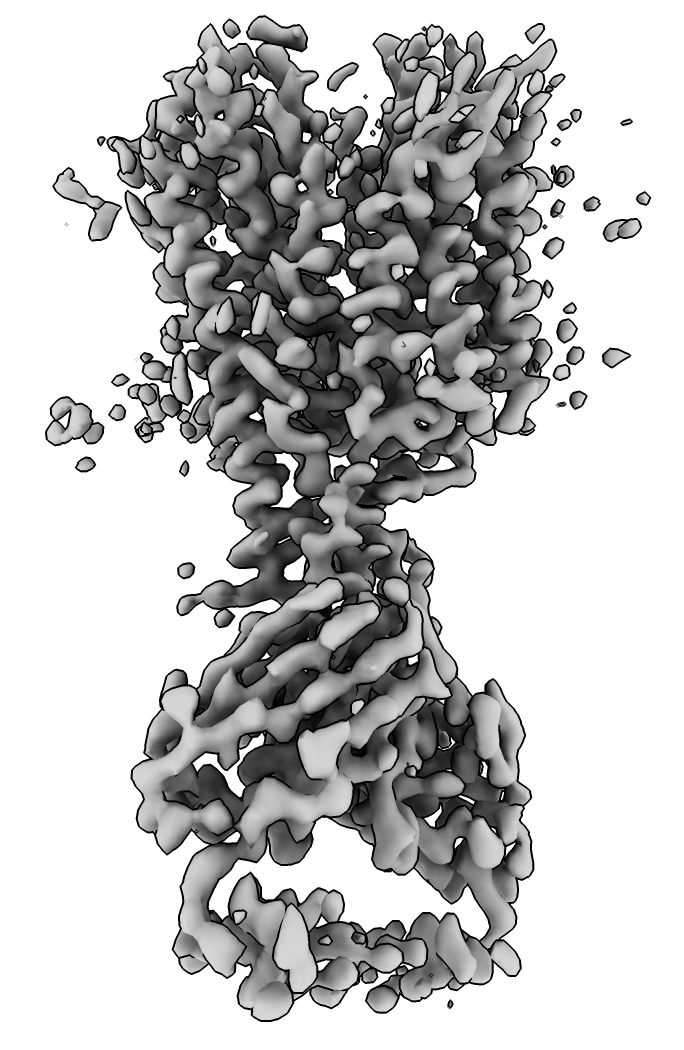
Now we look at another recently published cryoEM map and create a starting model by running AlphaFold since the protein is from chicken which is not one of the organisms in the AlphaFold database of precomputed structures. The EMDB map 23883 contains a membrane protein that transports omega-3 fatty acids and a bound antibody. We will model the transporter.
Structural basis of omega-3 fatty acid transport across the blood-brain barrier. Cater RJ, Chua GL, Erramilli SK, Keener JE, Choy BC, Tokarz P, Chin CF, Quek DQY, Kloss B, Pepe JG, Parisi G, Wong BH, Clarke OB, Marty MT, Kossiakoff AA, Khelashvili G, Silver DL, Mancia F. Nature. 2021 Jul;595(7866):315-319. doi: 10.1038/s41586-021-03650-9. PMID: 34135507; PMCID: PMC8266758.
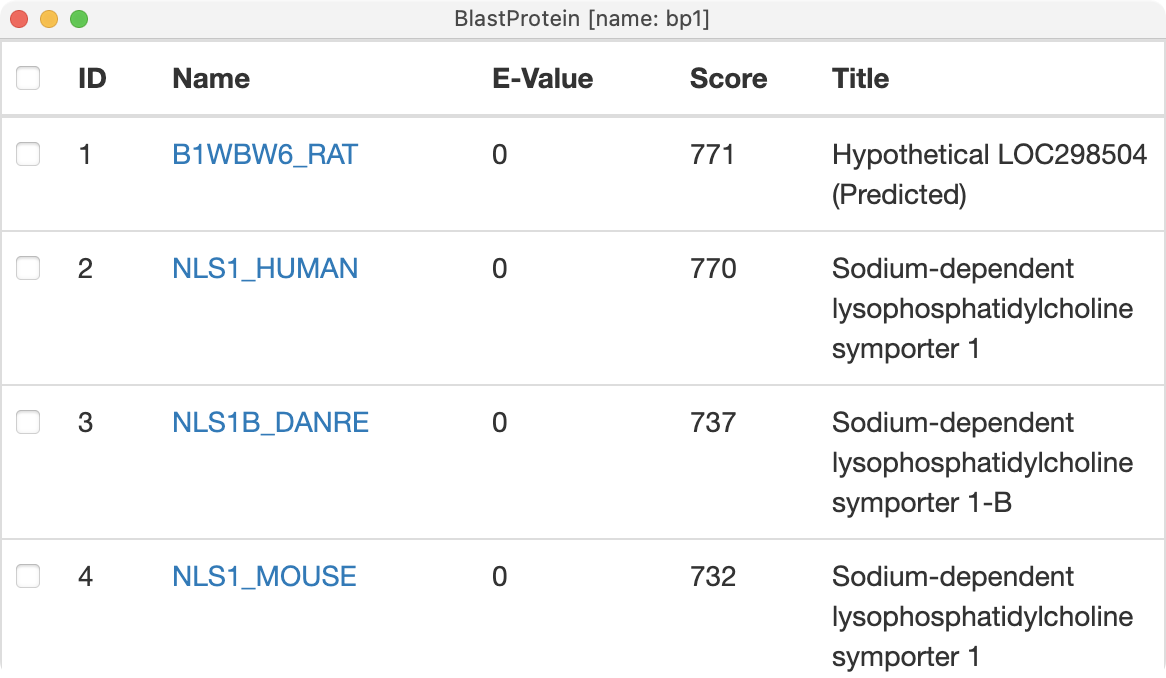
First lets take a look at what transporter homologs are available in the alphafold database. Paste the sequence, UniProt F1NCD6_CHICK into the ChimeraX AlphaFold panel (menu Tools / Structure Prediction / AlphaFold) and press the Search button to do a BLAST sequence search at 1e-3 E-value cutoff. Twenty one matches are found, the best being from rat, human, zebrafish, and mouse.
|  
| 
|
| EMDB map 23883. | AlphaFold database homologs to chicken sequence. | |
Click the rat, human and zebrafish links in the search output to load the AlphaFold structures. The Log panel reports that the sequences are 69% to 75% identical to the chicken sequence for our map.
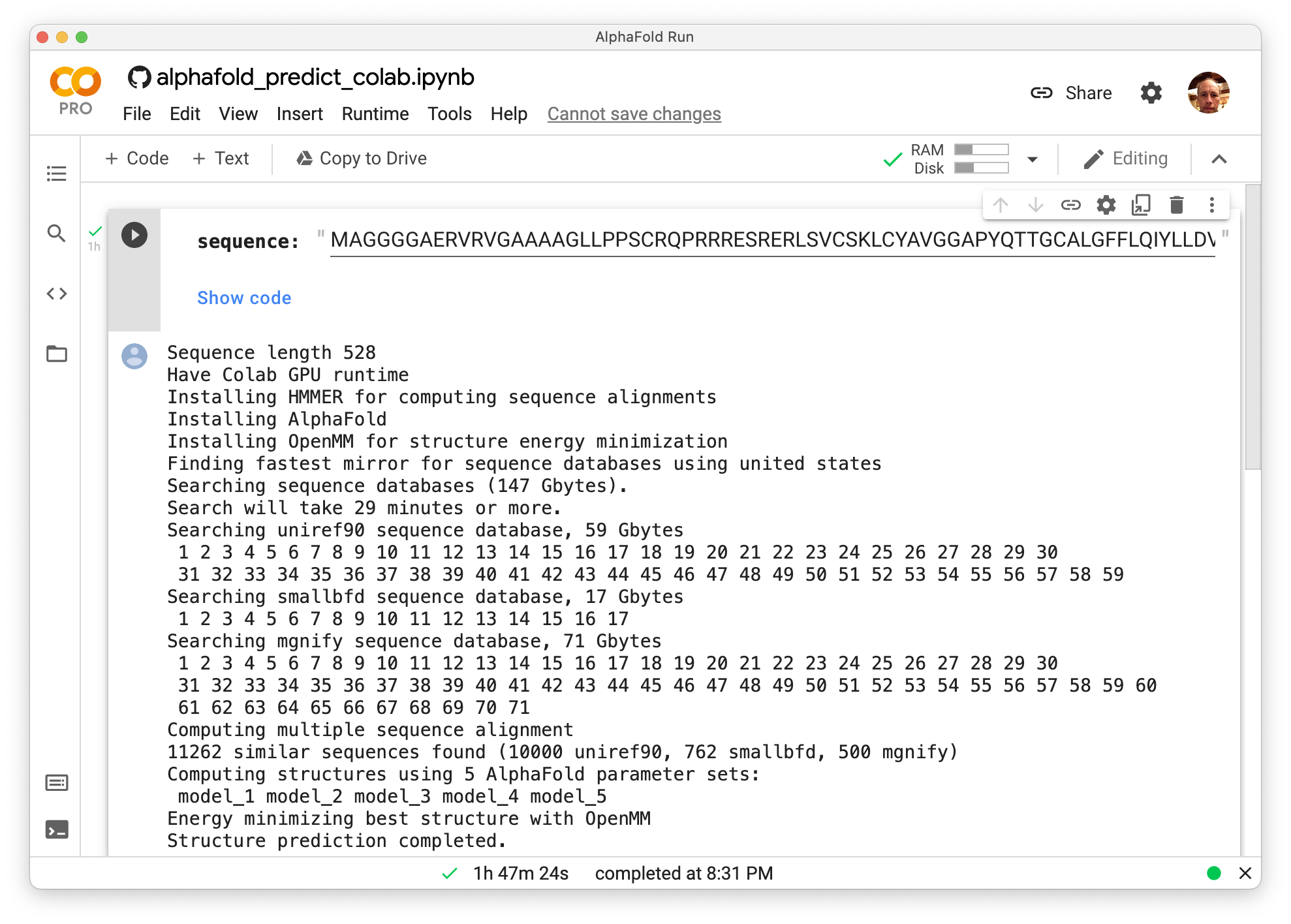
To predict the chicken sequence structure run AlphaFold by pressing the Predict button on the ChimeraX AlphaFold tool. This will run AlphaFold on Google Colab free servers. You will be asked to sign in to your Google account (same account used for Google email, drive, calendar). A security warning will display saying the ChimeraX AlphaFold code being run is not from Google, click Run Anyway.
The run took 1 hour 47 minutes to predict the 528 amino acid sequence. Log output shows that it installed HMMER (for computing a multiple sequence alignment), AlphaFold, and OpenMM (to energy minimize final structure), then searched 150 Gbytes of sequence databases (uniref90, smallbfd, mgnify), then ran the AlphaFold neural net with 5 alternative sets of parameters and selected the most confident resulting structure to energy minimize. The structure then was automatically loaded in ChimeraX. The best model is downloaded to your Downloads folder where ChimeraX keeps fetched files.
~/Downloads/ChimeraX/AlphaFold/prediction_13/best_model.pdb
| 
| 
|
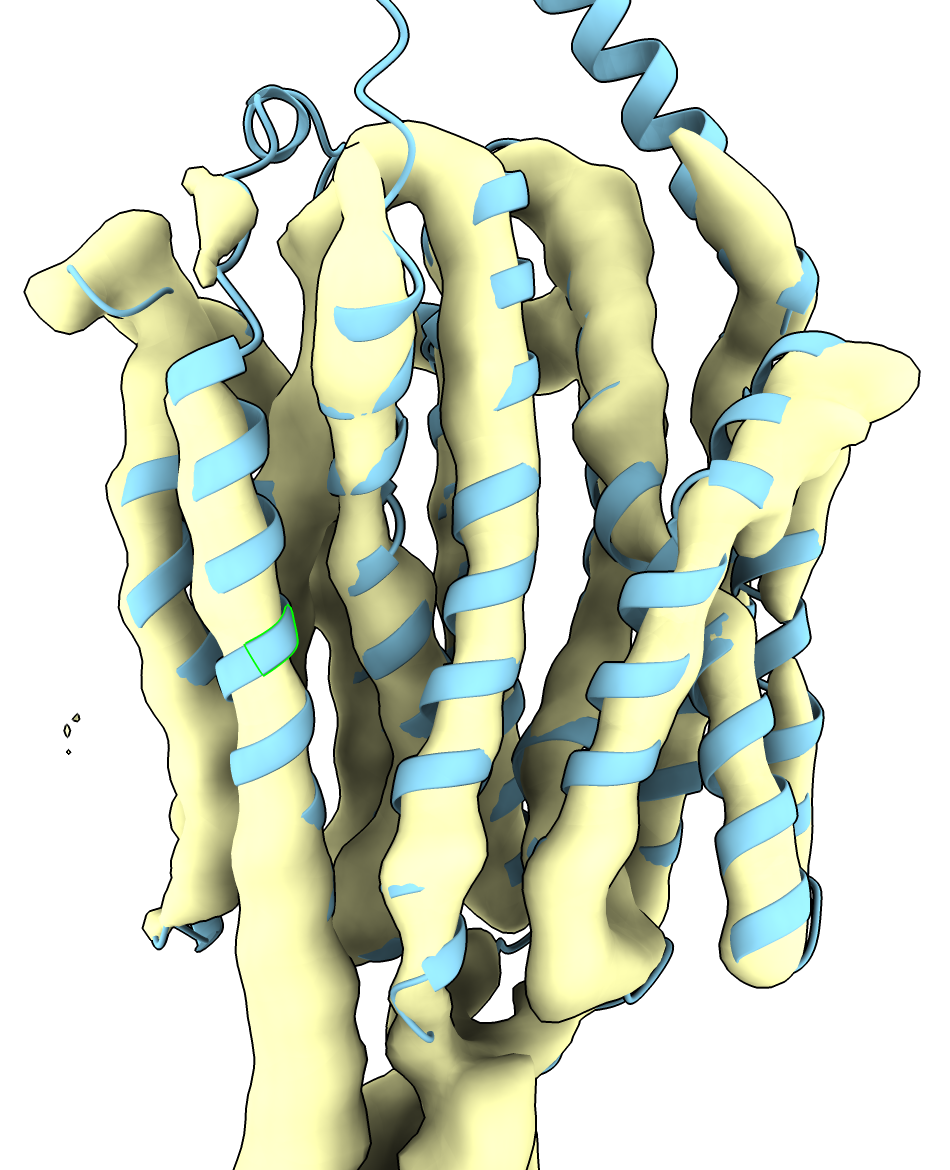
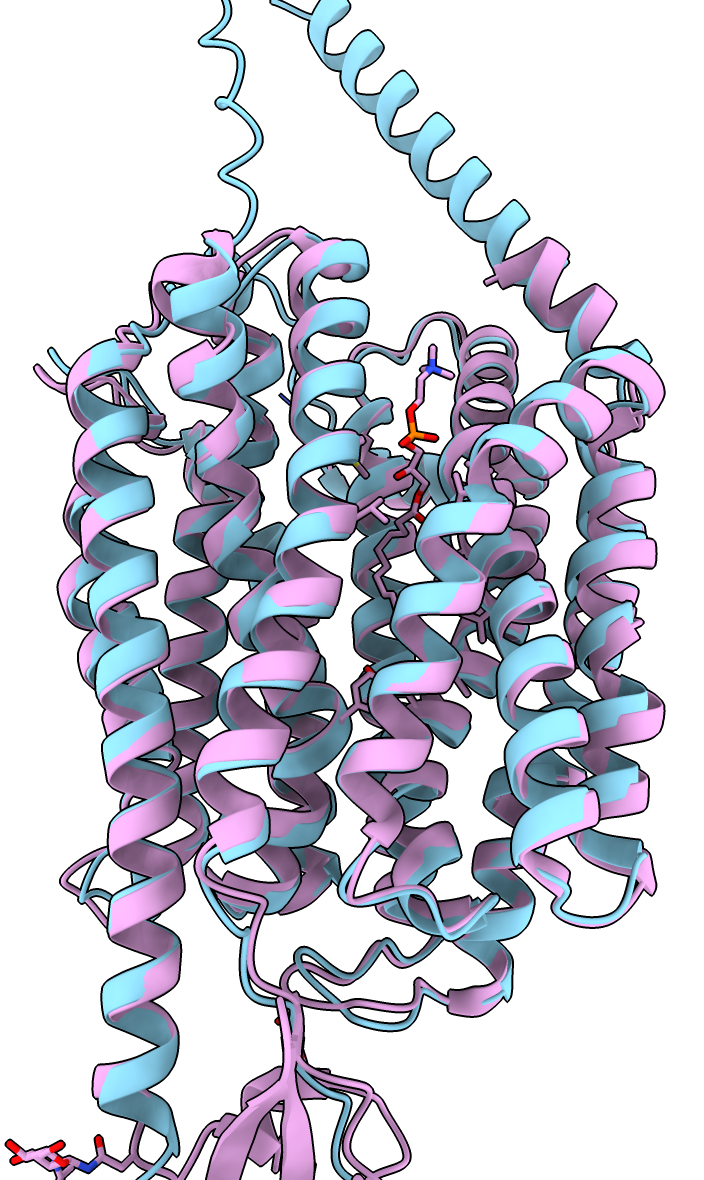
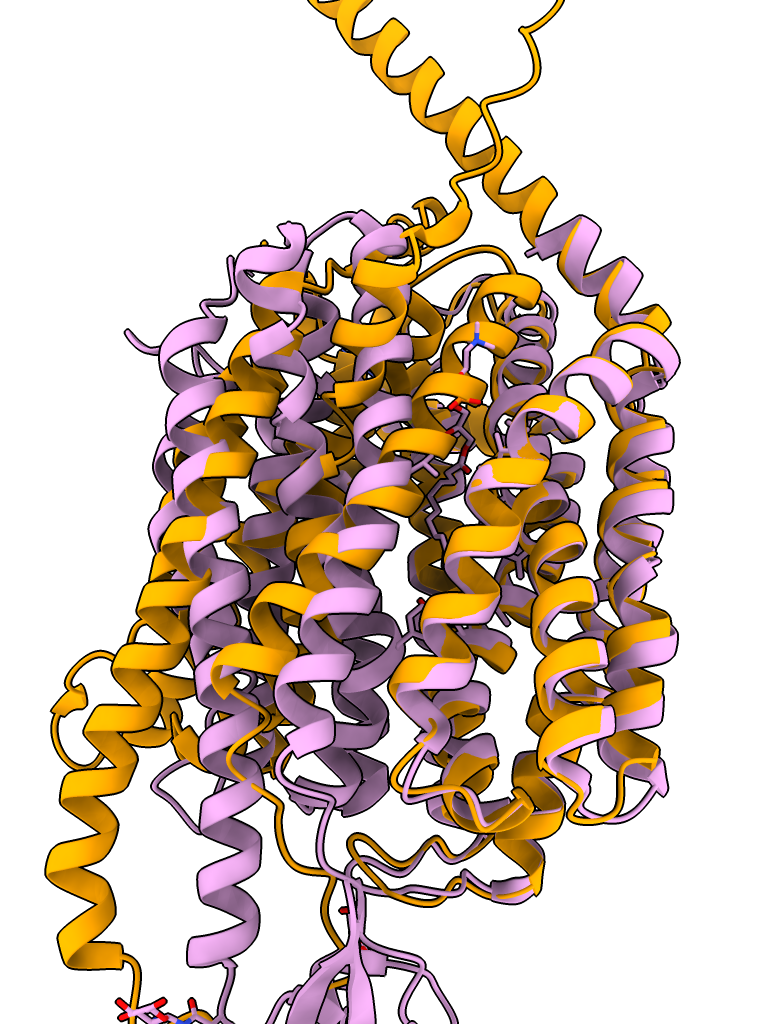
| Prediction compared to smoothed map. | Prediction (blue) compared to published cryoEM structure (pink). | AlphaFold rat homolog (orange) compared to cryoEM structure (pink). |
We fit the structure to the map using the Move Model mouse mode in the Right Mouse toolbar, and the Fit in Map panel from ChimeraX menu Tools / Volume Data / Fit in Map just as was done in the part 1 example. Using a smoothed map as computed with the volume gaussian #1 sdev 2 command as in part 1 helps visualize the fit agreement.
Comparing the predicted AlphaFold model to the structure solved by the authors of this data PDB 7MJS shows the helices agree extremely well.
The AlphaFold database homolog structures from rat, human and zebrafish all appear to be a different conformation of the transporter where several helices are in different positions, possibly a closed versus open state.
Time limits. The free Google Colab cloud servers only allow limited use per day, approximately 2 hours. That is only enough to compute one structure. To get more server time (up to 12 hours per day) Google offers Colab Pro for $10 per month.
Reduced sequence databases. AlphaFold uses reduced sequence database, 150 Gbytes versus about 2 Tbytes used for computing database entries and for the CASP14 structure prediction competition. This can reduce the quality of structures. AlphaFold works best with a multiple sequence alignment of hundreds to thousands of sequences.
No PDB structure templates. The Google Colab AlphaFold uses no PDB template structures.
Limited sequence length. The Google Colab AlphaFold calculation often gives out of memory or CUDA errors on sequences longer than 800 amino acids. We are studying this limitation and may be able to improve the ChimeraX AlphaFold script to increase the feasible sequence length.