Work in progress March 13 - May 22, 2025
Tom Goddard
May 22, 2025
Group meeting
Topics
- Boltz structure prediction tool
- Future plans for improving structure prediction
Boltz structure prediction
- Boltz is a structure prediction program derived from AlphaFold 3 with similar accuracy to AF3.
- Predicts assemblies of proteins, DNA, RNA, ligands, ions, solvent.
- Created by PhD candidate Jeremy Wohlwend in the Regina Barzilay lab at MIT.
- Fully open source, MIT license. AlphaFold 3, Chai-1 and other ML structure prediction programs
do not provide training code preventing the research community from improving the prediction method.
- bioRxiv preprint
Documentation
ChimeraX Boltz structure prediction tool
ChimeraX Boltz structure prediction tool will be in ChimeraX June release.
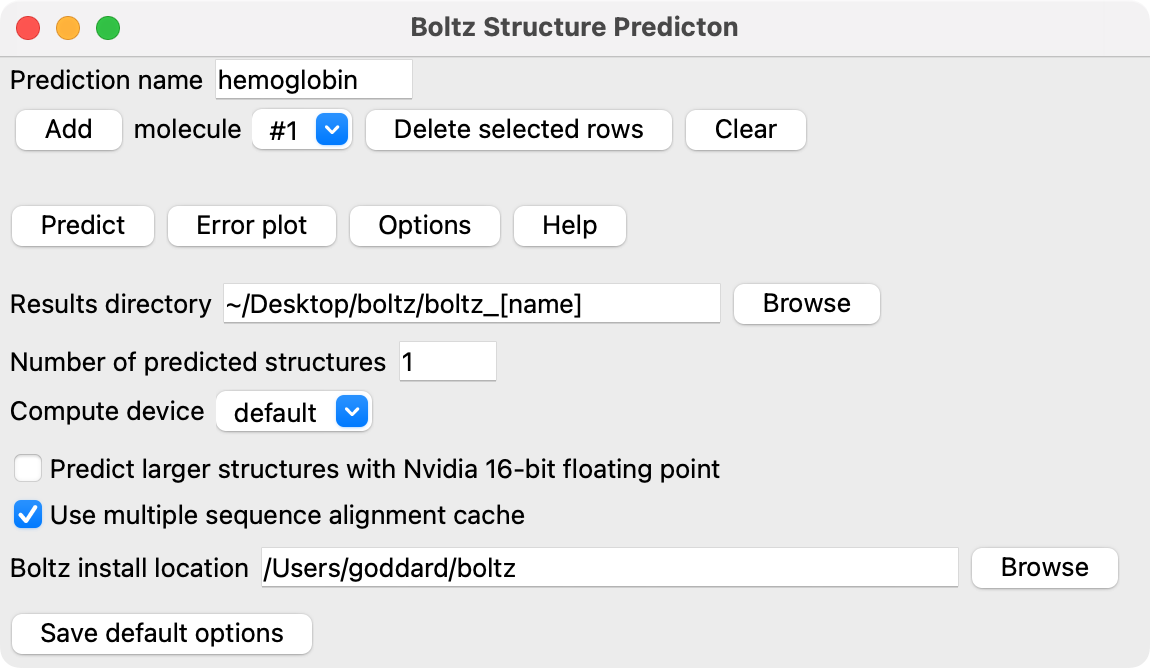
Boltz tool GUI
|
- New capabilities it adds:
- Predict structures with ligands and ions and nucleic acids.
- GUI to flexibly specify proteins, nucleic acids, ligands in various ways.
- Much faster than Colabfold / AlphaFold 2.
- Runs on local computer, one button install of Boltz on local machine (Mac, Windows, Linux).
- Easier specification of molecules to predict by using structures opened in ChimeraX.
- Implementation challenges:
- Estimate time for each job to plan predictions of large complexes that could take hours.
- Allow queueing many jobs to run sequentially and in background.
- Boltz uses Colabfold MSA server which is heavily loaded. May want UCSF MSA server.
- Boltz needs to read SDF format ligands to easily handle covalent linking, currently only CCD or SMILES.
Future Boltz improvements