Using AlphaFold protein structures in ChimeraX for cryoEM modeling
Tom Goddard
March 29, 2022
SBGrid webinar: Cryo-electron Microscopy of Membrane Proteins from Sample to Structure
AlphaFold capabilities in ChimeraX daily builds
- Fetch for best sequence match in AlphaFold database.
- Search with BLAST to find close sequences in AlphaFold database.
- Predict a multimer structure from sequences.
- Assess predicted errors to identify incorrect domain packing.
Finding a Starting Atomic Model for a cryoEM Map
We try to find an initial atomic model for the human
TACAN dimer structure
using the AlphaFold database
at the EBI
and ChimeraX. This map and an atomic model were published August 2021,
had no prior known homologous models in the Protein Databank
and is thought to be a mechano-sensitive ion channel involved
in pain sensation or lipid metabolism enzyme.
Cryo-EM structures of human TMEM120A and TMEM120B.
Ke M, Yu Y, Zhao C, Lai S, Su Q, Yuan W, Yang L, Deng D, Wu K, Zeng W, Geng J, Wu J, Yan Z.
Cell Discov. 2021 Aug 31;7(1):77. doi: 10.1038/s41421-021-00319-5. PMID: 34465718.
Fetch a structure from the AlphaFold Database
The AlphaFold database has about 1 million predicted structures (January 2022)
including all human genes, all genes from 20 model system organisms,
all SwissProt curated sequences, and sequences related to anti-microbial resistance
and neglected tropical diseases.
>sp|Q9BXJ8|TACAN_HUMAN Ion channel TACAN OS=Homo sapiens OX=9606 GN=TMEM120A PE=1 SV=1
MQPPPPGPLGDCLRDWEDLQQDFQNIQETHRLYRLKLEELTKLQNNCTSSITRQKKRLQE
LALALKKCKPSLPAEAEGAAQELENQMKERQGLFFDMEAYLPKKNGLYLSLVLGNVNVTL
LSKQAKFAYKDEYEKFKLYLTIILILISFTCRFLLNSRVTDAAFNFLLVWYYCTLTIRES
ILINNGSRIKGWWVFHHYVSTFLSGVMLTWPDGLMYQKFRNQFLSFSMYQSFVQFLQYYY
QSGCLYRLRALGERHTMDLTVEGFQSWMWRGLTFLLPFLFFGHFWQLFNALTLFNLAQDP
QCKEWQVLMCGFPFLLLFLGNFFTTLRVVHHKFHSQRHGSKKD
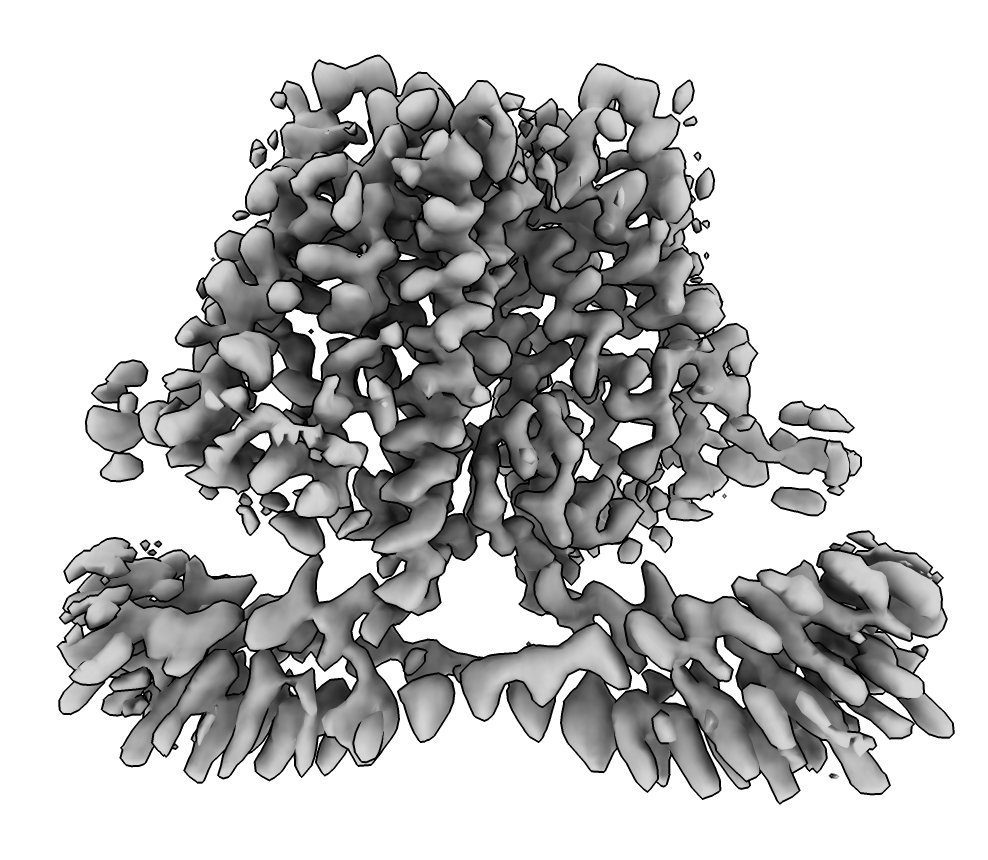
EMDB map 30495, 3.4 Angstroms.
(fetched with ChimeraX command open 30495 from emdb).
| 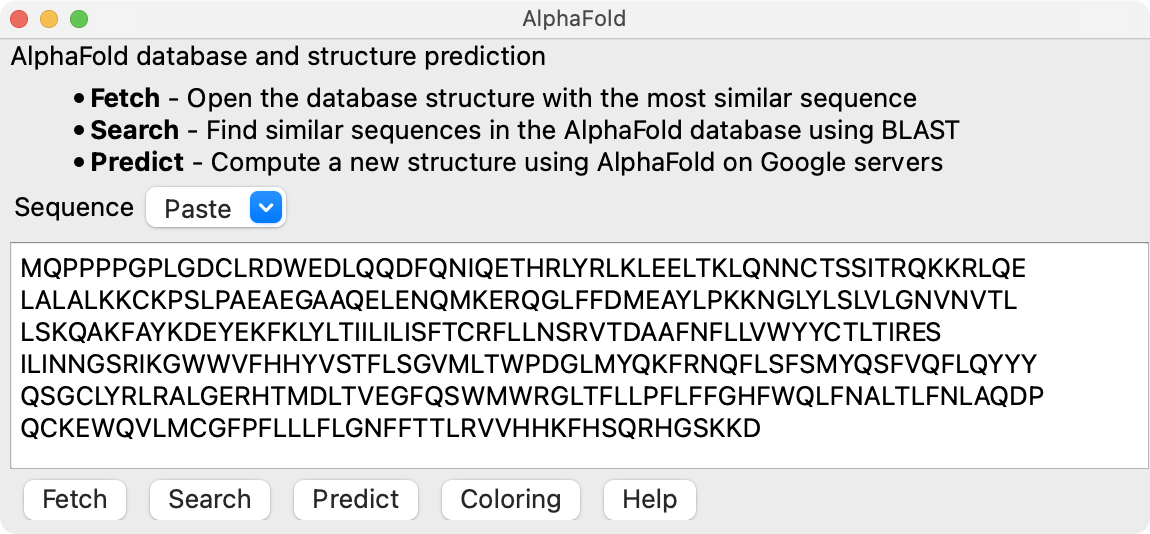
ChimeraX AlphaFold tool, in menu Tools / Structure Prediction,
with UniProt sequence TACAN_HUMAN,
then press Fetch button.
| 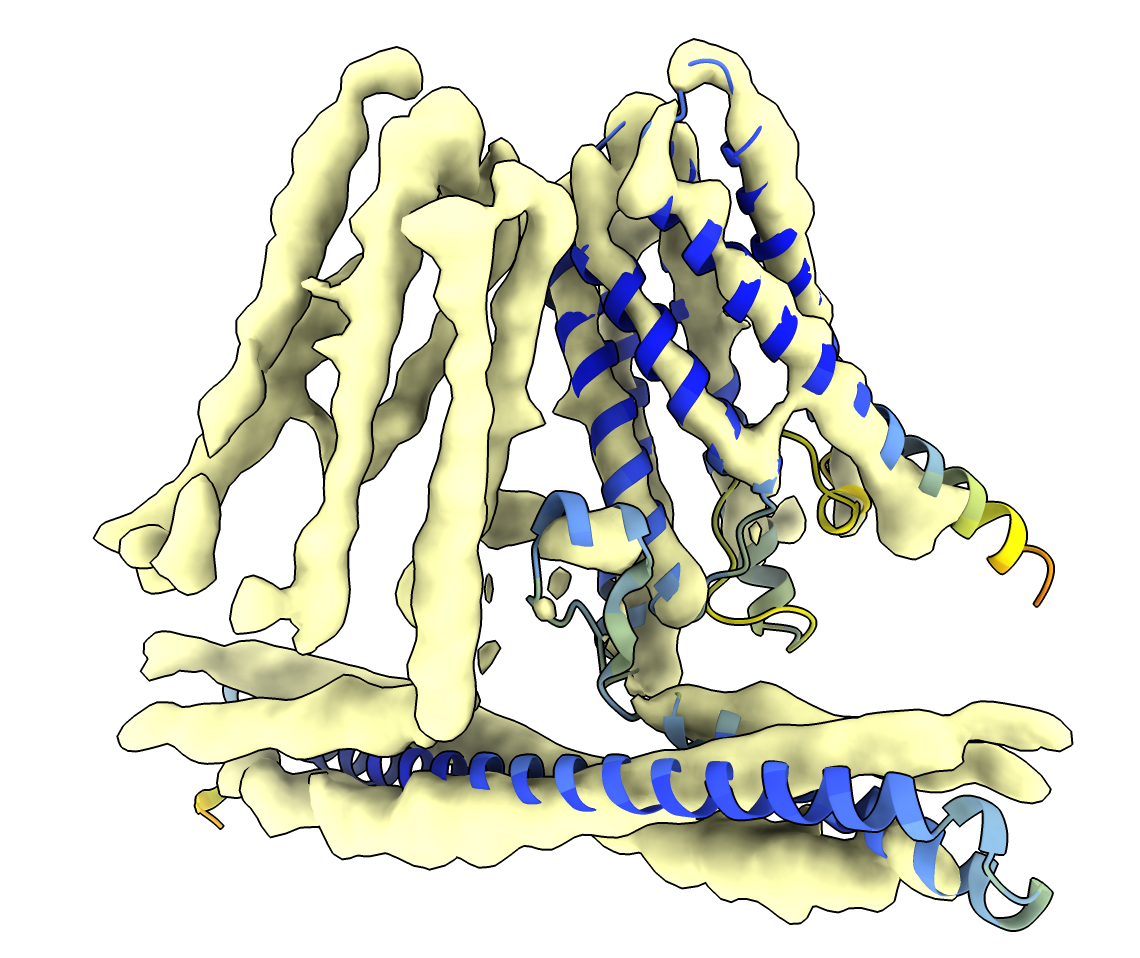
AlphaFold EBI database model fit into map (smoothed with volume gaussian #1 sdev 2).
|
The long intracellular alpha helix at the bottom can be rigidly moved with the ChimeraX
move atoms mouse mode to better fit the density to improve the initial model. Then
the atomic model can be refined in the map to correct side positions, e.g. with the ChimeraX
ISOLDE tool.
Searching the AlphaFold Database
Can find closest sequences in AlphaFold database with Search button.
This is useful to see different conformations when there is no close match.
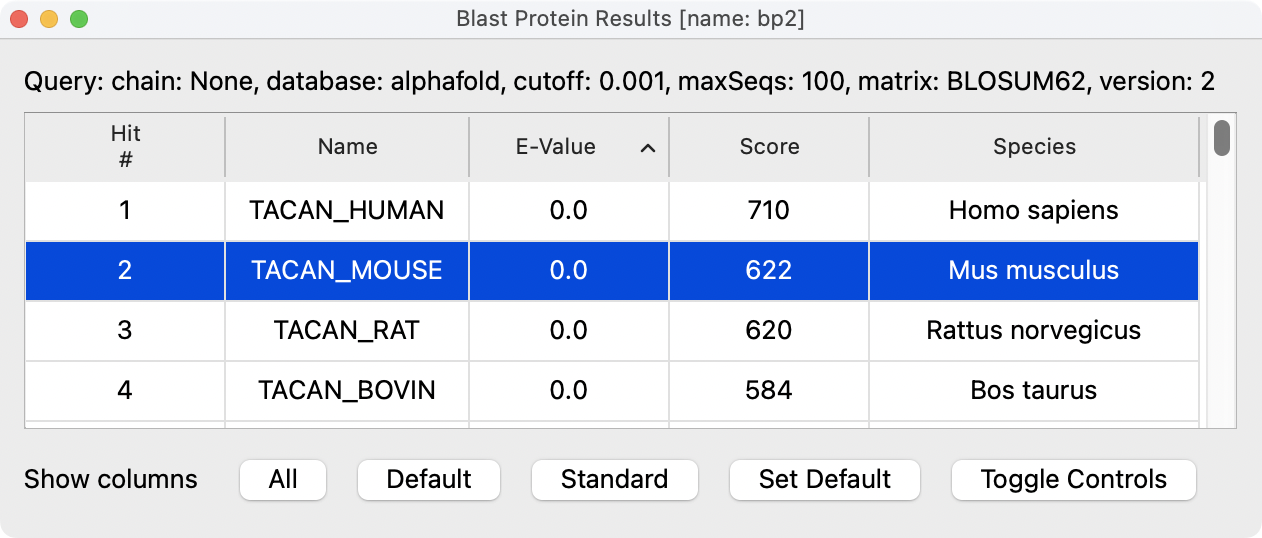
AlphaFold database BLAST search results for TACAN_HUMAN.
| 
Four best sequence matches in AlphaFold Database.
|
Running AlphaFold to Predict a Structure from a Sequence
AlphaFold database only has predictions of single proteins.
Use the Predict button to run AlphaFold on the TACAN dimer
after pasting two copies of the sequence separate by commas.
This will run AlphaFold on Google Colab free servers. You will be asked to sign in
to your Google account (same account used for Google email, drive, calendar).
A security warning will display saying the ChimeraX AlphaFold code being
run is not from Google, click Run Anyway.
Output
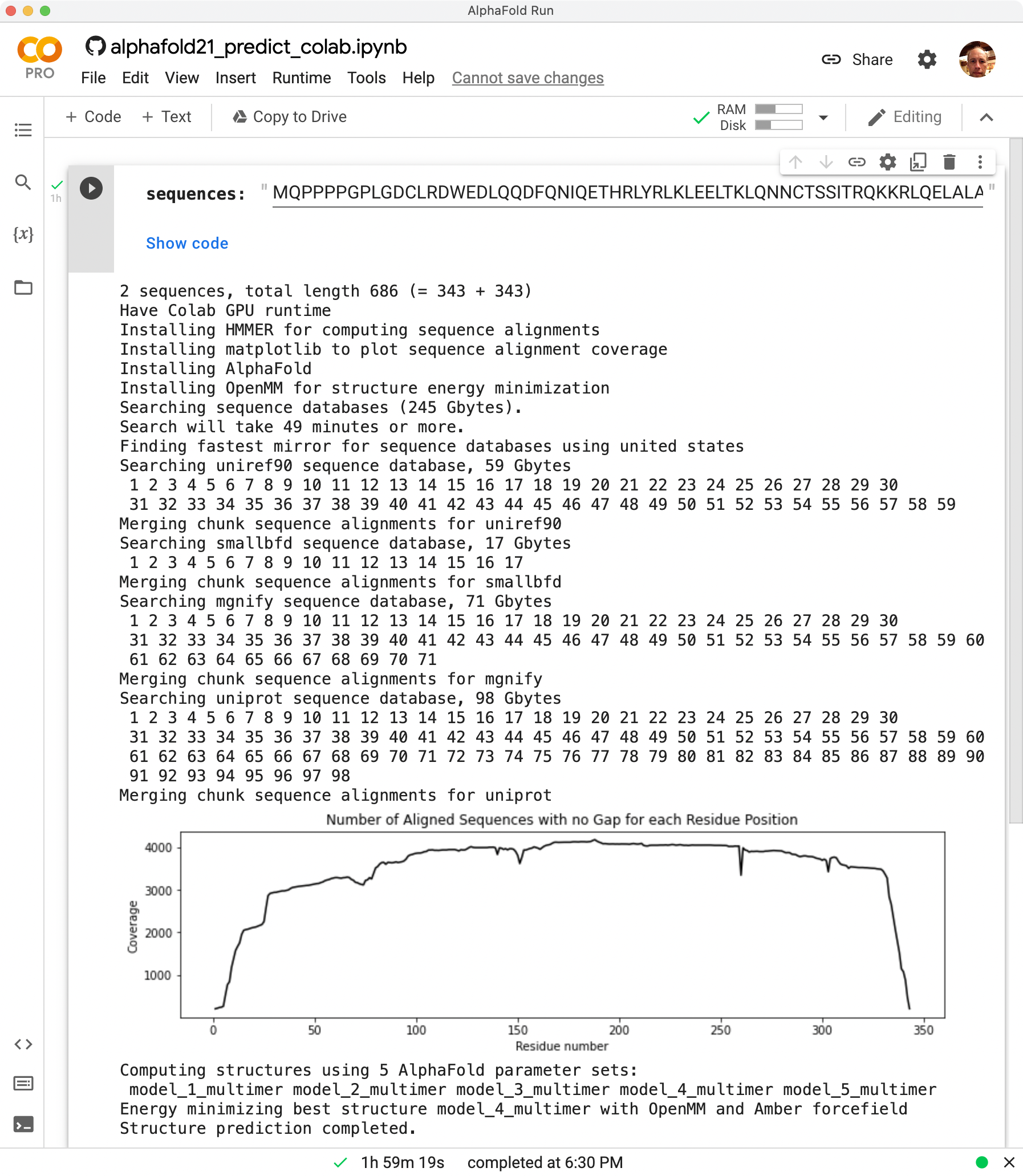
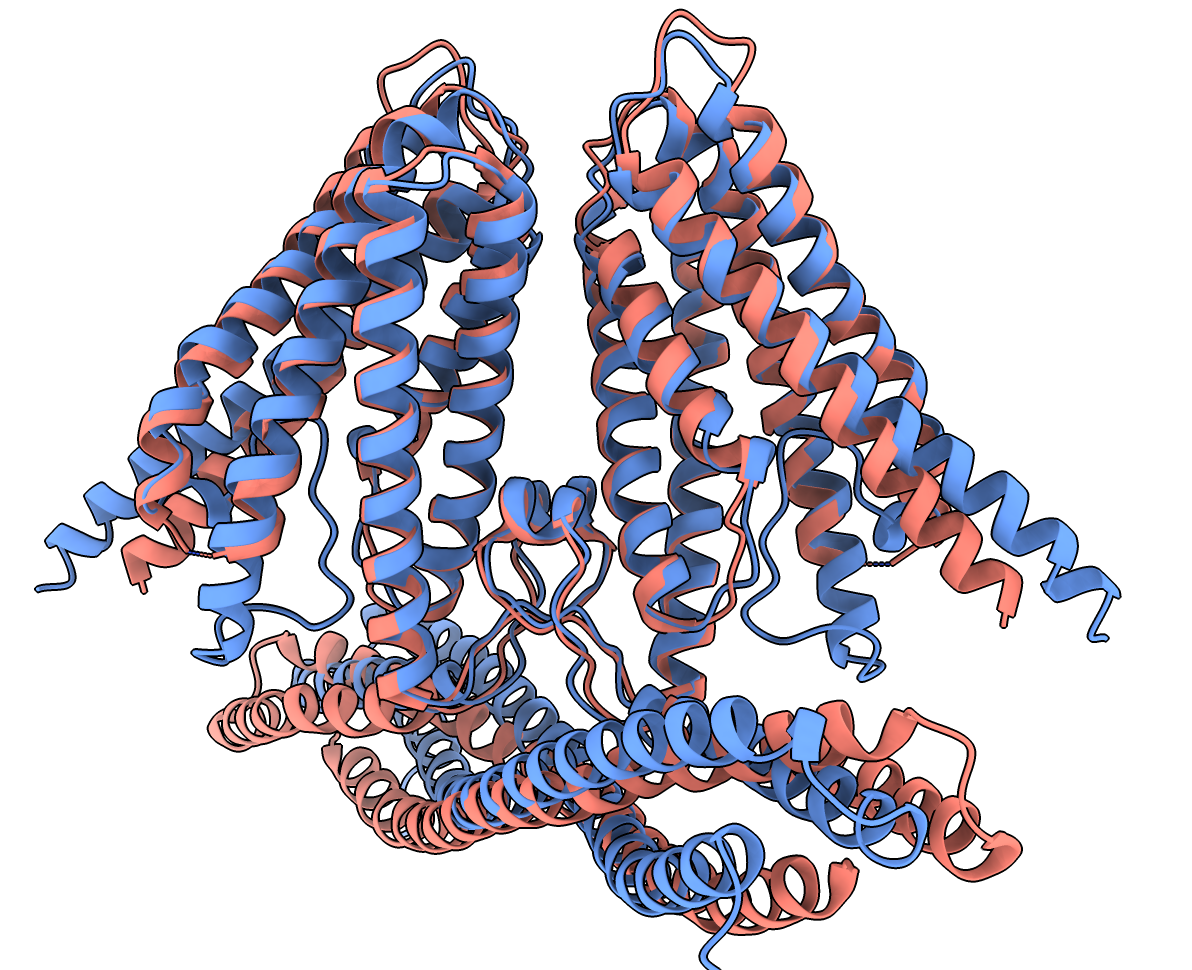
AlphaFold dimer (blue). Solved cryoEM structure 7cxr (red).
C-alpha RMSD 4 Angstroms.
- Took 2 hours to predict the 686 total amino acids.
- Started by installing software on Colab machine
- HMMER for computing a multiple sequence alignment.
- AlphaFold to predict structure.
- OpenMM to energy minimize final structure.
- Searched 250 Gbytes of sequence databases:
uniref90, smallbfd, mgnify, uniprot.
- Ran the AlphaFold neural net with 5 different sets of parameters.
- Energy minimized the highest scoring structure.
- Results automatically downloaded including the
best model.
~/Downloads/ChimeraX/AlphaFold/prediction_3
AlphaFold Predicted Aligned Error (PAE)
How do you judge if an AlphaFold model is correct without a known structure?
- pLDDT confidence score for each residue (0-100 range), 50 = low confidence colored red, 90 = high confidence colored blue.
- Predicted Aligned Error (PAE) for relative alignment for every pair of residues, distance in Angstroms (0-35 range).
Predicted Aligned Error is shown with Error Plot button on ChimeraX AlphaFold panel.
Residues can be clustered that have low PAE to define domains with the Color PAE Domains
button. Different domains may be misaligned relative to one another.
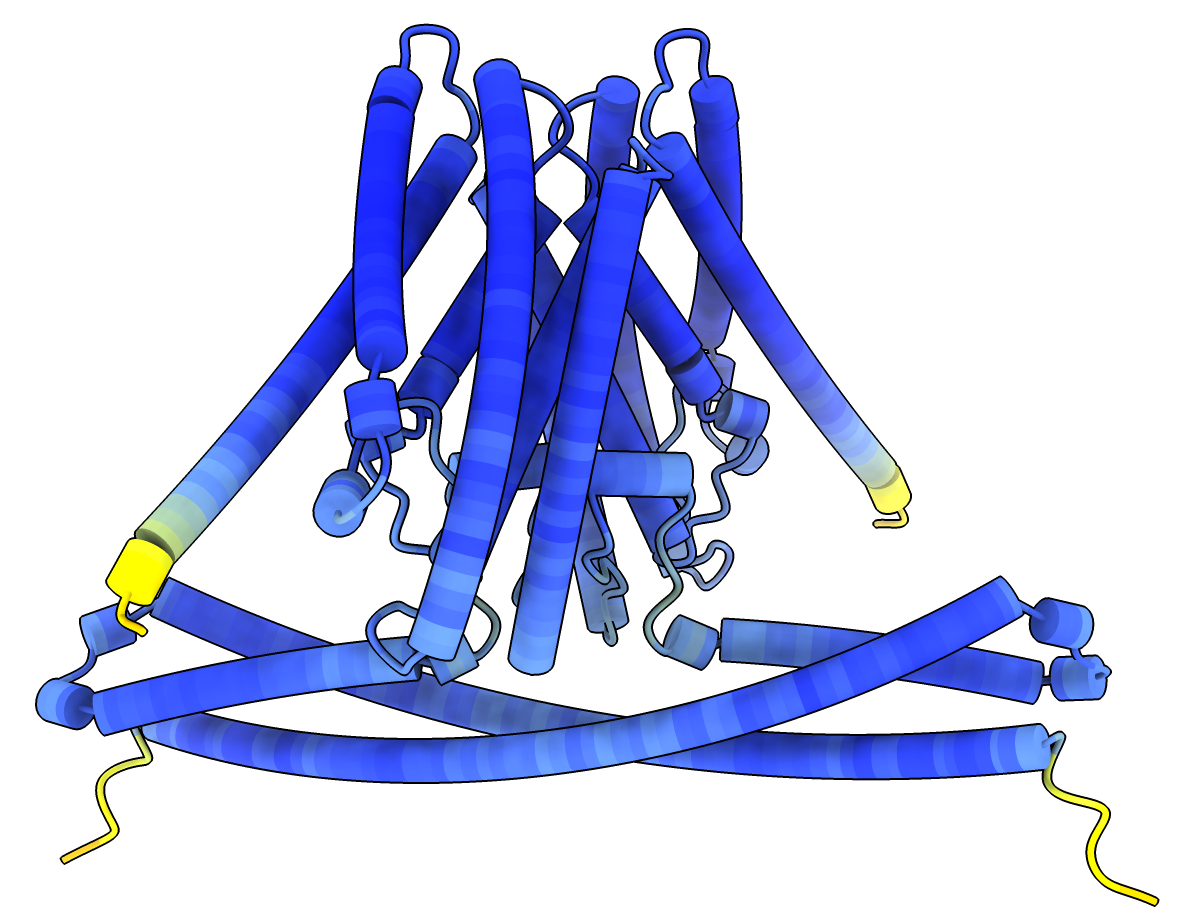
pLDDT per-residue confidence coloring.
| 

Residue vs residue PAE values.
| 
Domains with high PAE between them may be incorrectly packed.
|
Limitations of AlphaFold
- Maximum sequence length
~1000 amino acids for Colab AlphaFold predictions due to limited GPU memory (16 Gbytes).
- Only handles 20 standard amino acids.
- Does not handle ligands, ions, solvent, nucleic acids....
- Predictions take hours.
- About 1/3 of interfaces are wrong in multimer predictions.
Running AlphaFold on your own computer
With a modern GPU (NVidia RTX 3090 24 Gbytes, A40 48 Gbytes) not available on Colab
you can predict structures of 2000 or 3000 amino acids.
- Needs GPU with lots of memory ($2000 - $10000) for better performance than Colab.
- Requires large databases, 2 Tbytes, that can takes a long time to download.
- Speed for sequence alignment is improved with databases on fast SSD drive.
- Requires Linux.