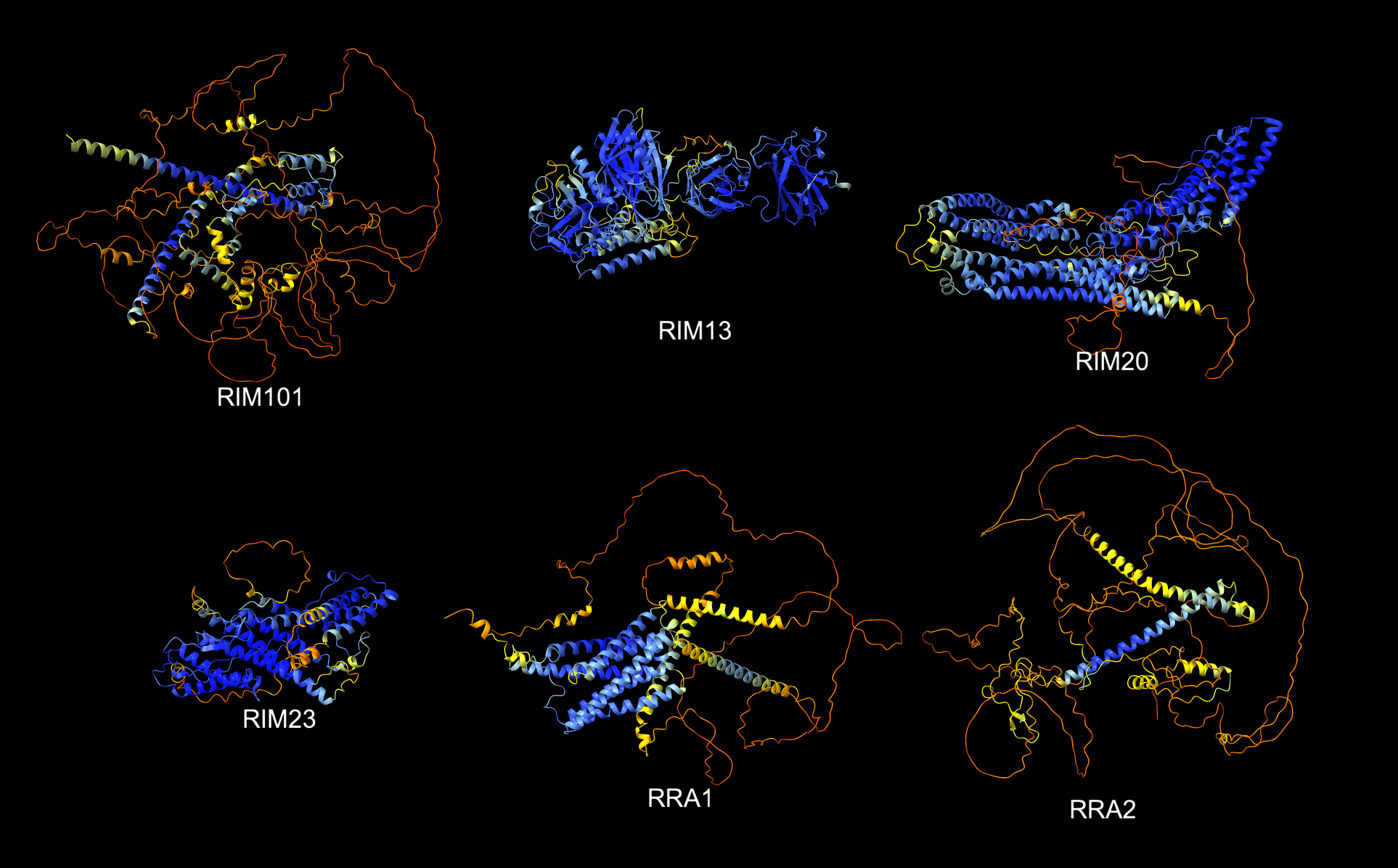
colored by pLDDT score, blue confident, red not confident.
Tom Goddard
goddard@sonic.net
January 31, 2024
We look at how to use AlphaFold to find which proteins bind to each other among a set of tens or hundreds of proteins. The procedure is to predict dimers for all protein pairs and look for ones where AlphaFold has high confidence in the binding interface. I've added some commands to ChimeraX to simplify running and evaluating these AlphaFold predictions. They are in ChimeraX versions newer than January 31, 2024.
Hiten Madhani at UCSF is interested in the RIM pathway in Crytococcus neoformans which senses extracelluar pH. Little is known about the interactions of these 6 proteins RIM101, RIM13, RIM20, RIM23, RRA1, RRA2. Sequences rim.fasta
|
|
| AlphaFold predictions for 6 RIM monomers colored by pLDDT score, blue confident, red not confident. |
First we'll predict the 6 monomer structures using AlphaFold. Make a directory for the predictions
$ mkdir ~/rim_monomers $ mv ~/Downloads/rim.fasta ~/rim_monomers
The following ChimeraX commands estimate the predictions will take about 1 hour.
cd ~/rim_monomers
alphafold monomers rim.fasta
Estimated prediction time 1 hour 9 minutes using Nvidia 3090 GPU.
6 monomer sequences with lengths 488-916:
RIM101 (916), RIM13 (813), RIM20 (902),
RIM23 (488), RRA1 (654), RRA2 (743)
Prediction command: colabfold_batch sequences.fasta .
Run these predictions from a Linux shell. ColabFold will write file log.txt showing progress and create many files rim_monomers.zip (50MB, list) including 5 structures for each sequence.
$ colabfold_batch rim.fasta . >& colabfold.out &
Display the structures using ChimeraX command
alphafold monomers open .
Now predict the 21 dimers using AlphaFold. Make a new directory for the predictions
$ mkdir ~/rim_dimers $ mv ~/Downloads/rim.fasta ~/rim_dimers
The following ChimeraX commands estimate the predictions will take about 16 hours.
cd ~/rim_dimers
alphafold dimera rim.fasta output rim_dimers.fasta
21 dimers with lengths 976-1832.
Estimated prediction time 16 hours using Nvidia 3090 GPU.
Prediction command: colabfold_batch --num-recycle 3 rim_dimers.fasta .
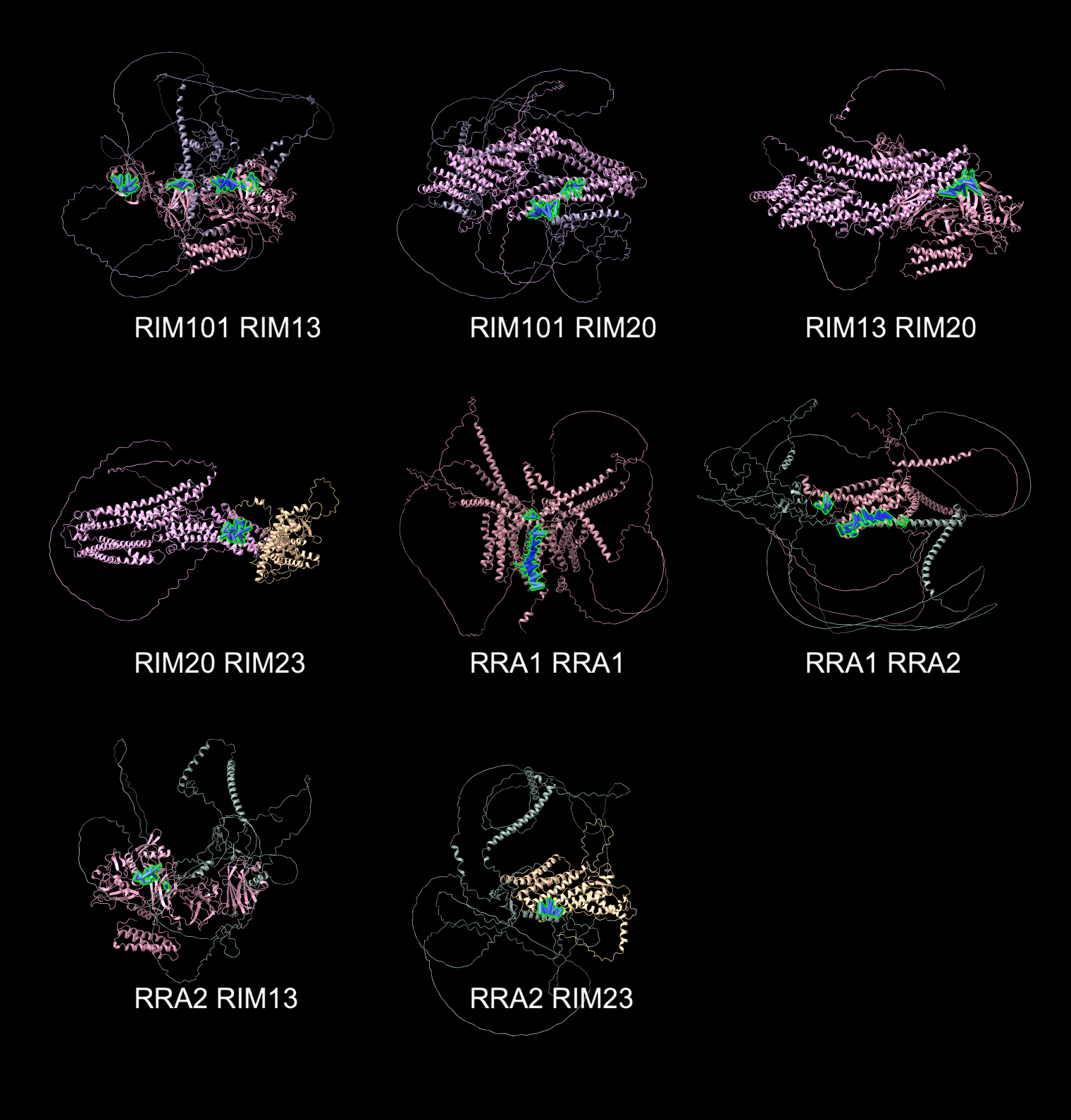
|
| 8 predicted dimers with binding interface (green) having high AlphaFold confidence (PAE <= 5A for at least 10 close residue pairs. |
Run these predictions using the rim_dimers.fasta file from a Linux shell producing log.txt and many files rim_dimers.zip (500 MB, list) including 5 structures for each dimer.
$ nohup colabfold_batch --num-recycle 3 rim_dimers.fasta . >& colabfold.out &
ChimeraX can check every predicted dimer (21 * 5 = 105 models) to see if AlphaFold assigns high confidence to the binding interface using ChimeraX command
alphafold interfaces .
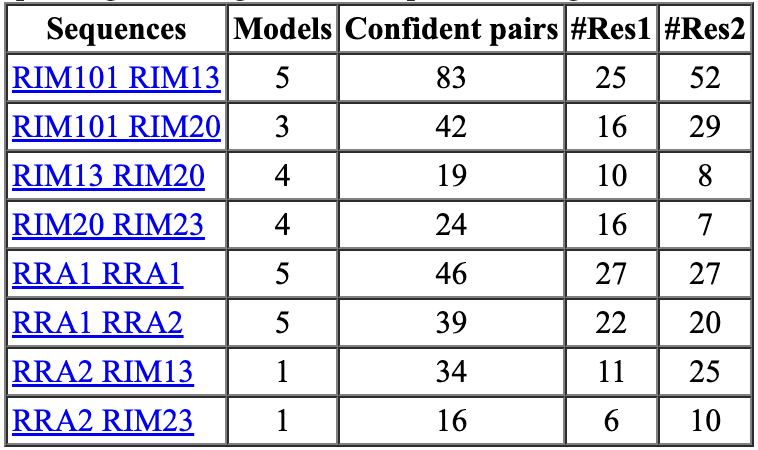
8 of 21 dimers have 10 or more confident residue interactions
spanning <= 4 Angstroms with predicted aligned error <= 5 Angstroms.
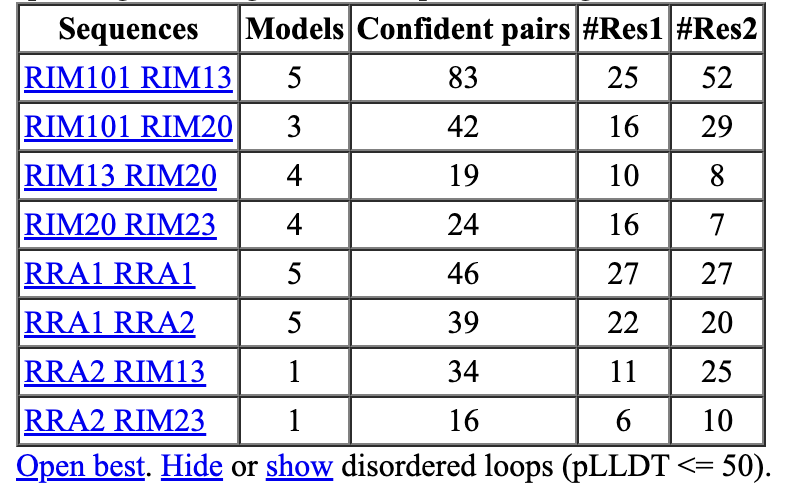
ChimeraX shows a table of dimers where AlphaFold assigns high confidence to the binding interface. Clicking Open best link shows the predicted dimers.
Are any of these RIM dimer predictions correct? We don not know. Different colorings can show AlphaFold predicted errors.
Predicted local distance different test (pLDDT) is a score (0-100) assigned to each residue measuring whether AlphaFold is confident that the residue C-alpha to neighbor residue C-alpha distances would match the true structure.
Predicted aligned error (PAE) is a score for each pair of residues assessing if one residue was aligned with the true structure, how far off would the second residue be from its location in the true structure, measured in Angstroms (0-35).
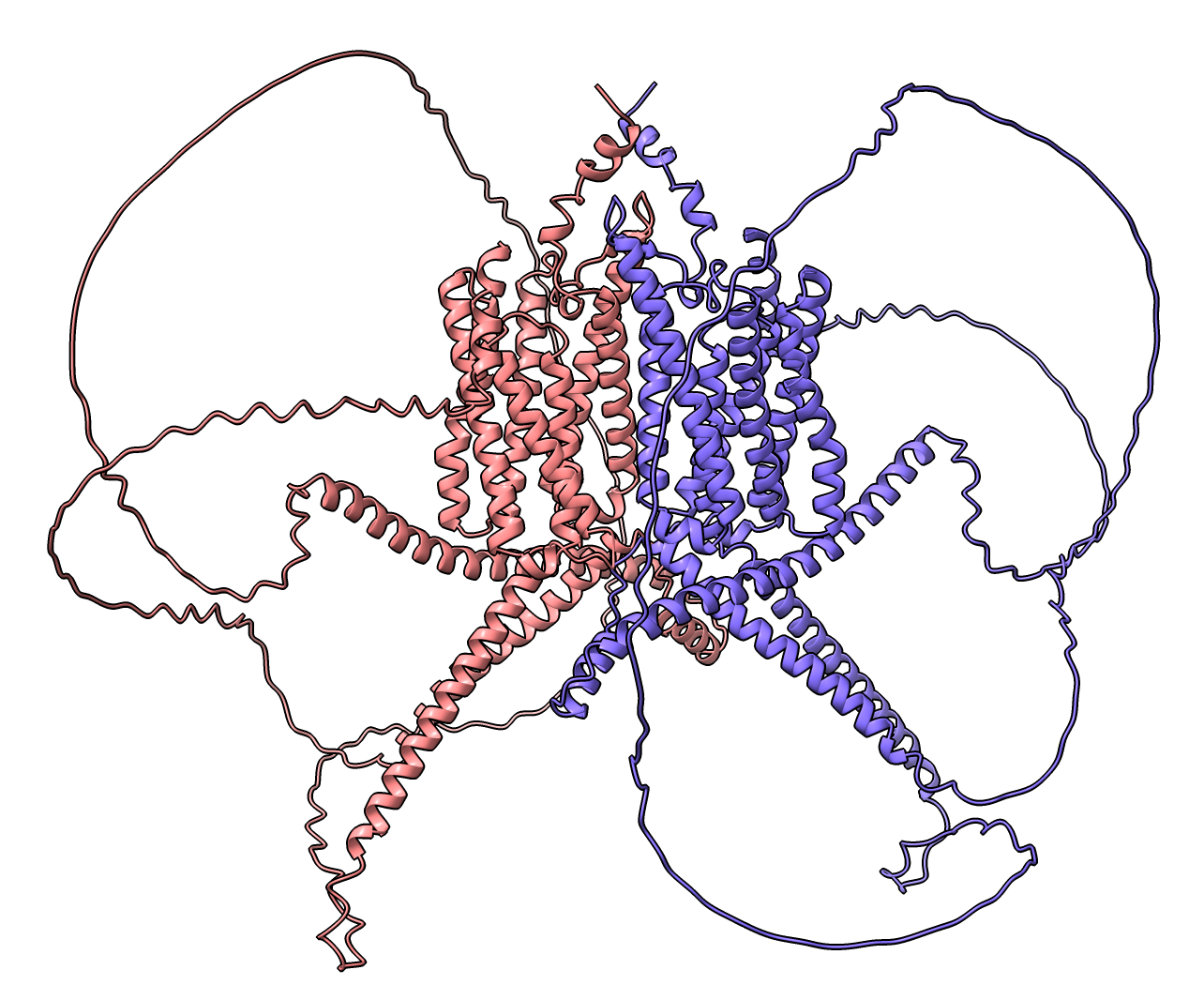
RRA1 RRA1 dimer prediction. Command: color #1 bychain or press Chain Coloring button in Molecule Display toolbar. | 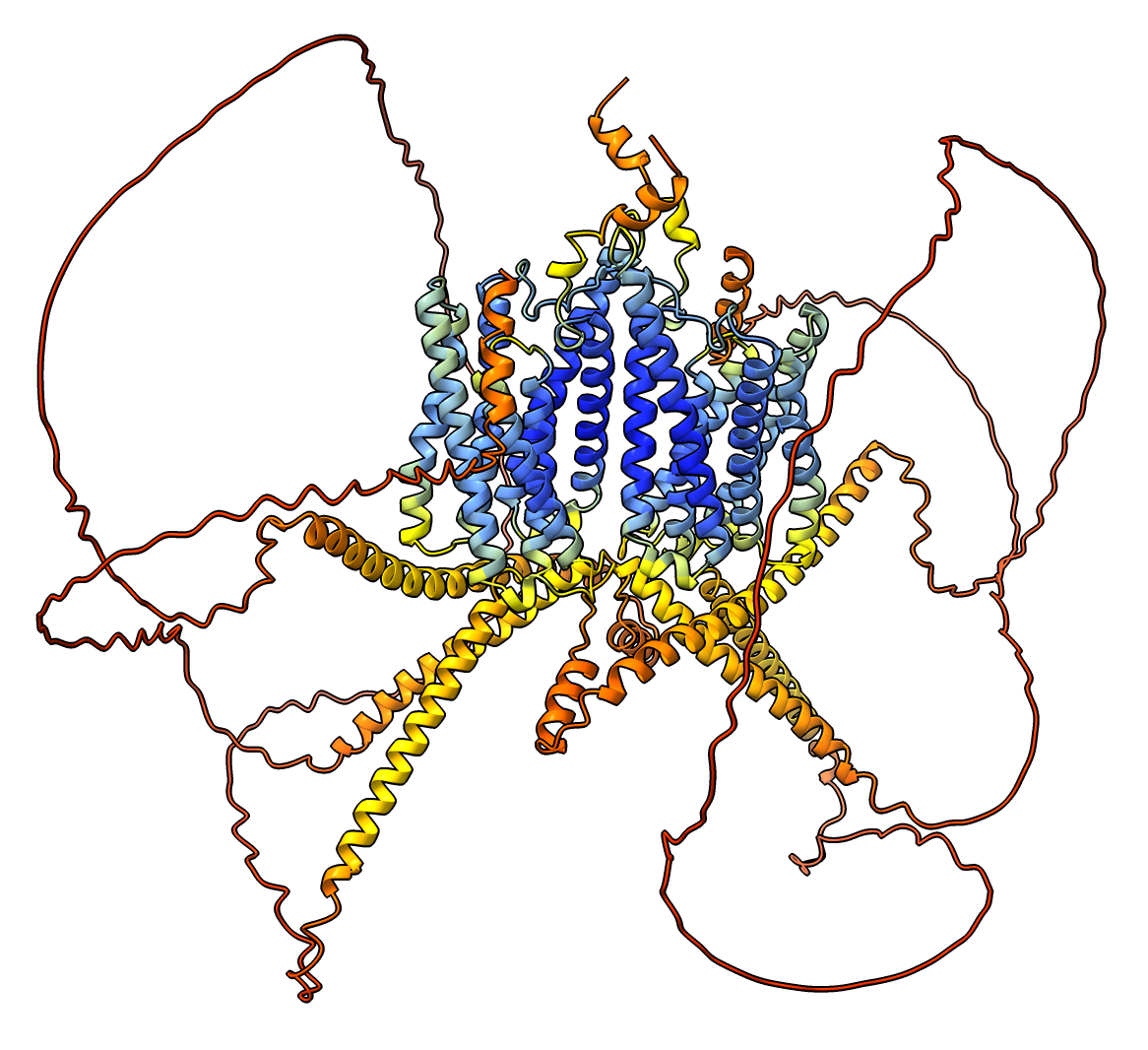
Predicted local distance difference test score (pLDDT), blue high confidence. Command color bfactor #1 palette alphafold or press Color pLDDT button on PAE plot. | 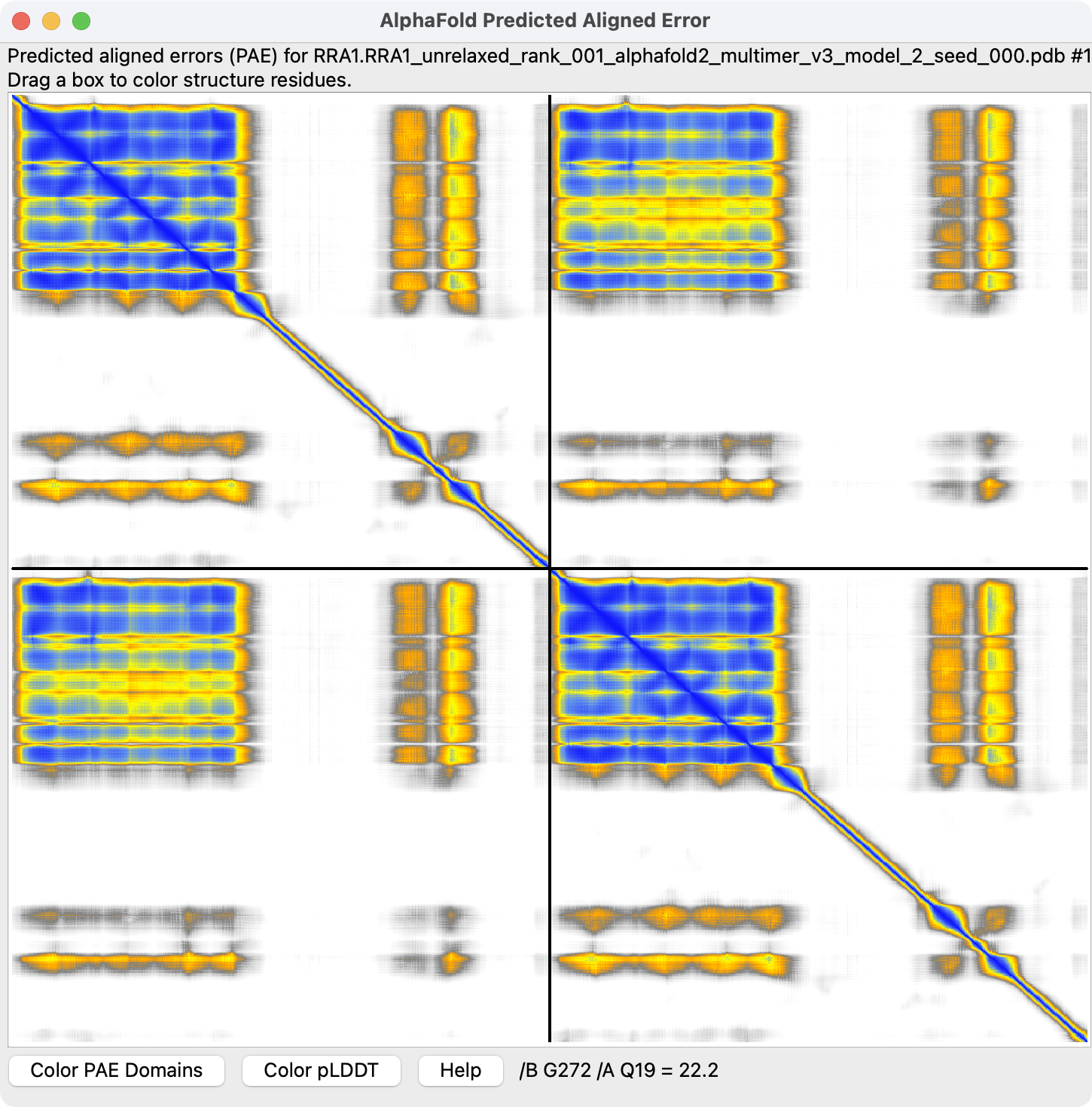
Predicted aligned error heat map, confidence in the relative orientation of each pair of residues, both axes are residue number. Show plot with menu Tools / Structure Prediction / AlphaFold Error Plot. |
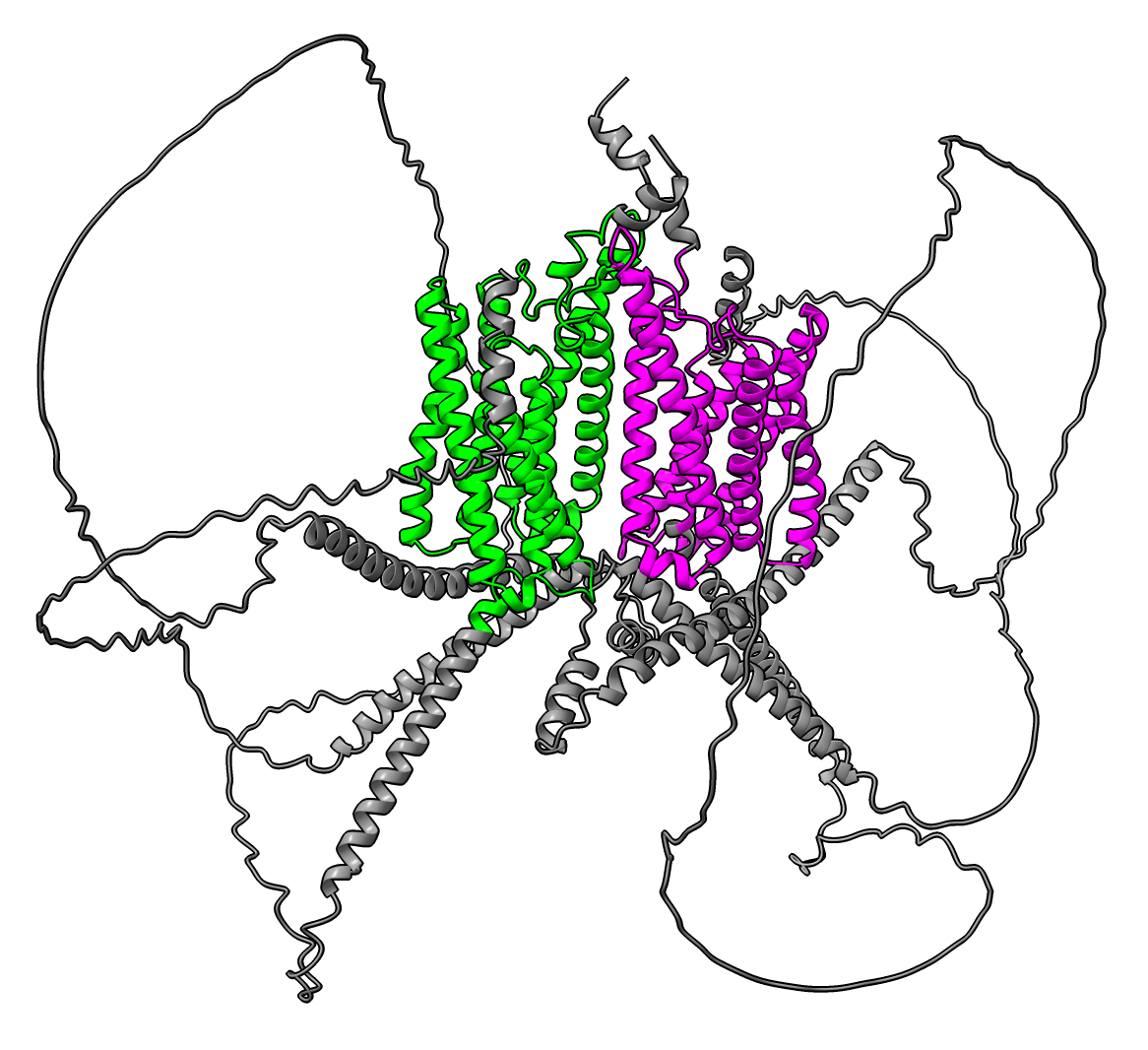
Dragging box around off diagonal blue/yellow square in PAE heatmap colors the two domains having confident relative orientation. | 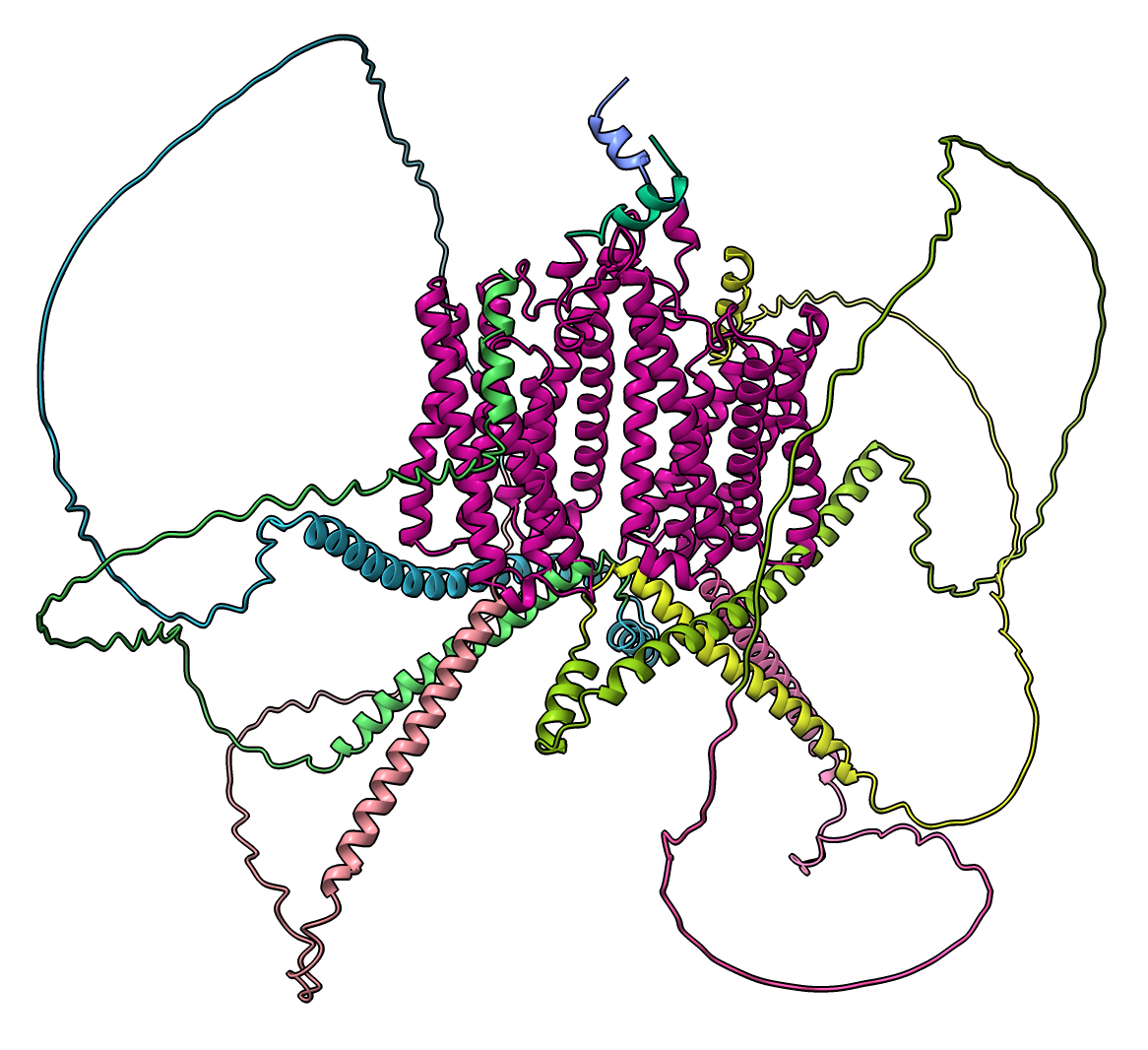
Predicted aligned error domains. Residues confidently aligned to each other are grouped together. Command: alphafold pae #1 colorDomains true or press Color PAE Domains button on PAE plot. | 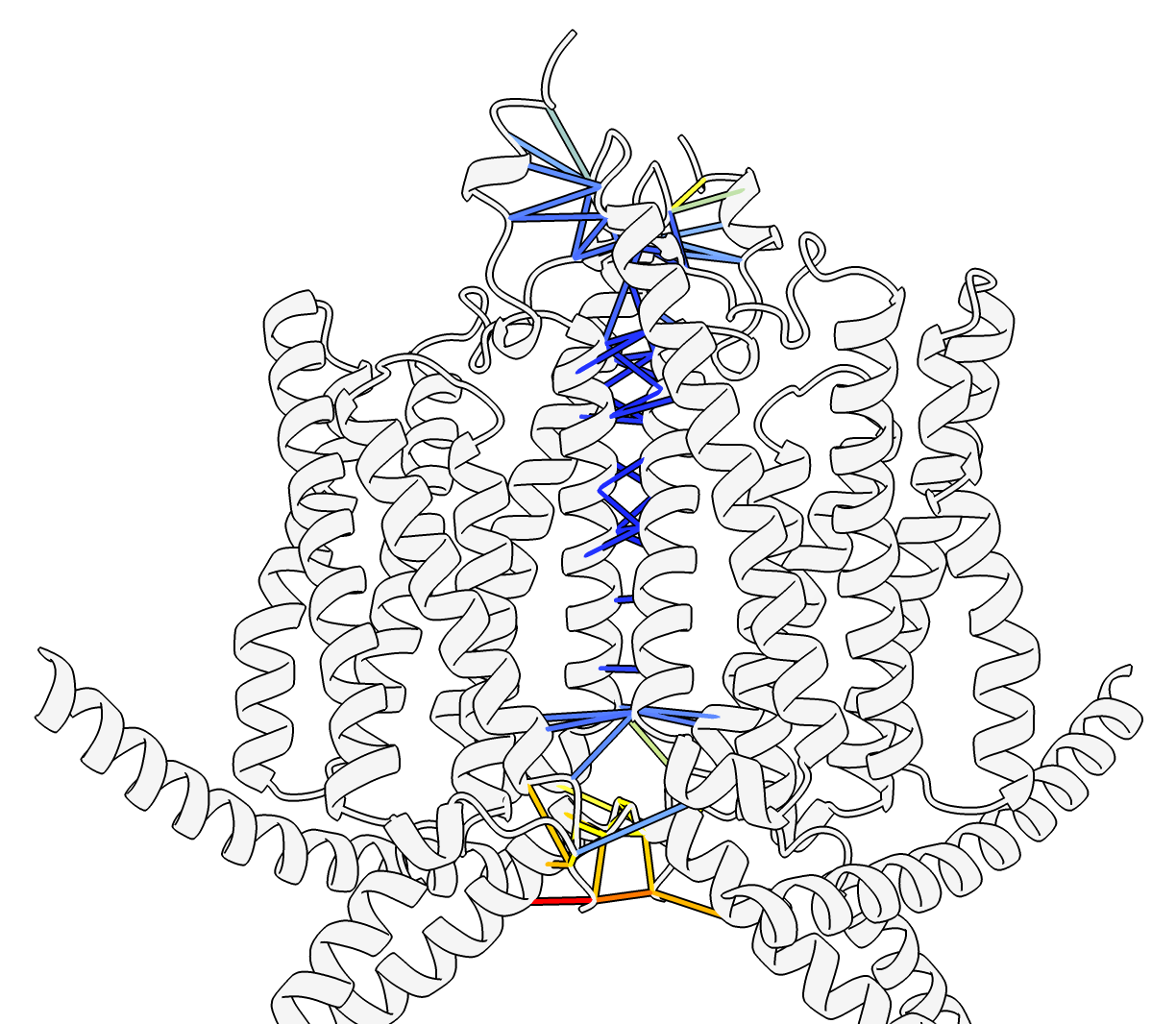
Predicted aligned error (PAE), blue high confidence. Command: color #1 lightgray ribbon Command: alphafold contacts #1/A to #1/B distance 4 |
5 models superimposed. Command: matchmaker #2-5 to #1 | 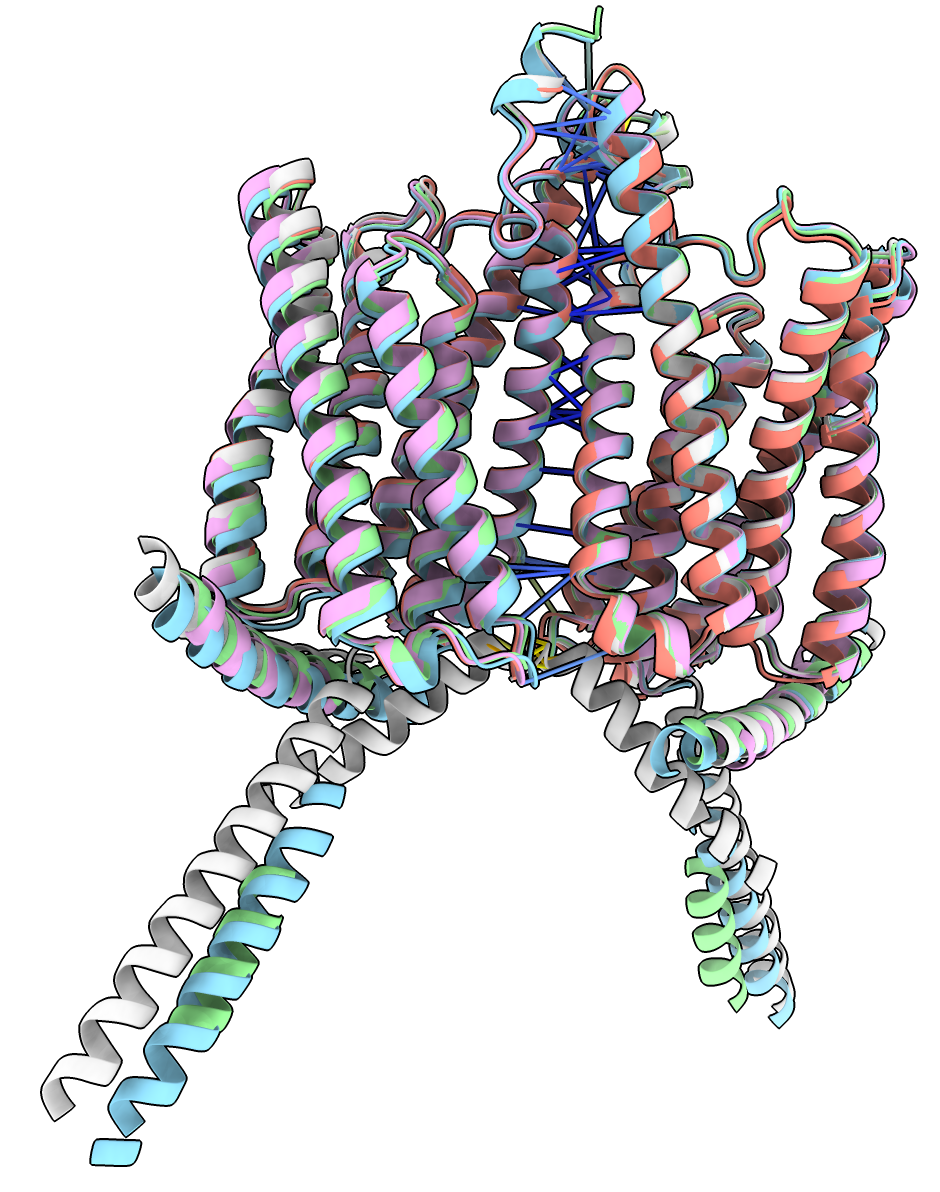
5 models superimposed only pLDDT > 50 shown. Command: hide @@bfactor<50 ribbon |
We use ColabFold which is an optimized version of AlphaFold that runs 5-10 times faster. I installed localcolabfold on a Linux computer (Ubuntu 22.04, quillian.cgl.ucsf.edu) with an Nvidia 3090 GPU. Using Linux and an Nvidia GPU is the only way to make fast predictions. Predictions with just a CPU are possible but they are 60 times slower.
Yes. Here are instructions. It requires two steps because the Wynton compute nodes do not have internet access, and ColabFold uses a cloud-based sequence alignment server to make the deep sequence alignment required by AlphaFold.
To work around this the colabfold_batch command can be run twice. The first time is to compute the sequence alignments and must be run on the Wynton login node that has internet access
colabfold_batch --msa-only dimers.fasta >& colabfold_seqs.out &
The second time it is submitted to the GPU queue and run without the --msa-only flag.
About 200 dimer predictions can run in a week on a single Nvidia 3090 GPU assuming sizes uniformly between 500 and 2500 residues. The runtime increases as the square of the total number of residues in the dimer.
The total number of residues of a predicted structure is limited by the GPU memory. With 24 GBytes on the Nvidia 3090 the maximum size is about 3000 residues. With an Nvidia A40 with 48 Gbytes I have run predictions up to 4700 residues. Running a prediction with too many residues leads to out of memory, or other GPU errors.
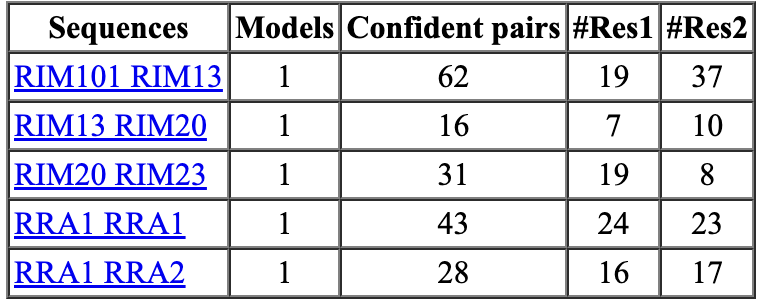
The standard AlphaFold or ColabFold prediction produces 5 models using 5 differently trained neural networks. By predicting only 1 model with 1 chosen network it can run 5 times faster. Also the number of recycles (iterations through the network) can be decreased from the default 3 to 1 making it 2 times faster (decreases the cycles from 4 to 2). These two changes can make predictions 10 times faster. The RIM example runs in 1.5 hours instead of 14 hours on a single Nvidia 3090 GPU with these settings but only finds 5 protein-protein interfaces instead of 8.
Another approach is to use a faster GPU. On an Nvidia 3090 the default 5 models and 3 recycles took 14 hours, and on an Nvidia A40 (48 GB) it took 16 hours. An Nvidia 4090, A100, or H100 would probably be able to make the predictions 1.5 to 2 times faster judging by benchmarks comparing those processors to an Nvidia 3090.
The prediction time increases with the square of the number of residues. For N residues the prediction time is approximatel (N/30)*(N/30) seconds. So predicting smaller structures will increase the prediction speed considerably.
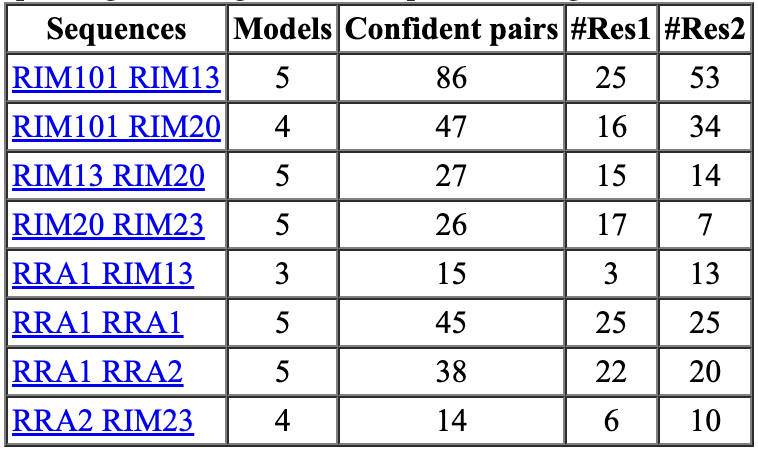
A study claims that increasing the number of recycles AlphaFold parameter from 3 to 20 gives better protein-protein interface predictions. But the prediction time is increased as much as a factor of 5. For our RIM case using 20 recycles took 69 hours compared to 3 recycles taking 14 hours. 20 recycles also found 8 protein-protein interactions one of which was a pair of proteins not found with 3 recycles.
| Accurate 20 recycles, 69 hours | Default 3 recycles, 14 hours | Fast 1 recycle, 1 model, 1.5 hours |
|---|---|---|
| 
| 
|