Making OpenFold Structure Predictions in ChimeraX
Tom Goddard
February 11, 2026
OpenFold in ChimeraX
ChimeraX can run the OpenFold 3 structure prediction method
to compute atomic structures of proteins and nucleic acids, including modified residues, ligands and ions
on your laptop or desktop computer.
A ChimeraX graphical user interface (menu Tools / Structure Prediction / OpenFold)
and ChimeraX command (openfold) are provided to make predictions.
ChimeraX daily builds dated February 13, 2026 and newer use OpenFold 3 preview.
OpenFold 3 is fully open source under the Apache 2.0 license.
OpenFold installation
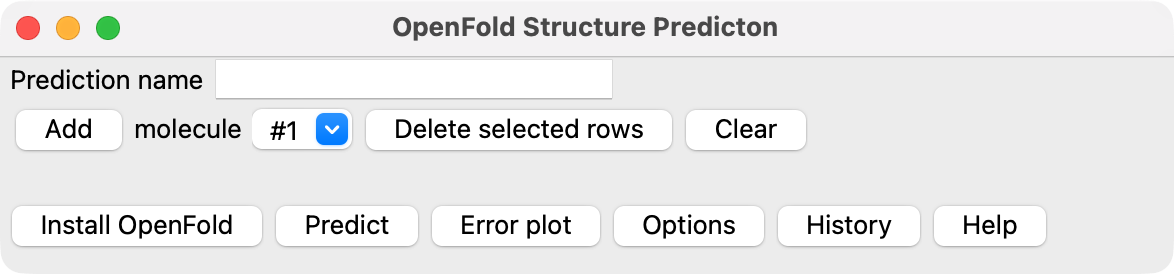
When you first start the OpenFold tool within ChimeraX (menu Tools / Structure Prediction / OpenFold) it will
show a button Install OpenFold. OpenFold is a large software package, taking about 1 Gbyte of disk space
and uses the Torch machine learning package. It also requires the neural network weights (2 Gbytes)
to make predictions. Downloading and installing can take 10 minutes or more depending on network speed
OpenFold will be installed in your home directory in ~/openfold3 and the network weights in ~/.openfold3.
The ChimeraX openfold install command can also be used to do this one time installation.
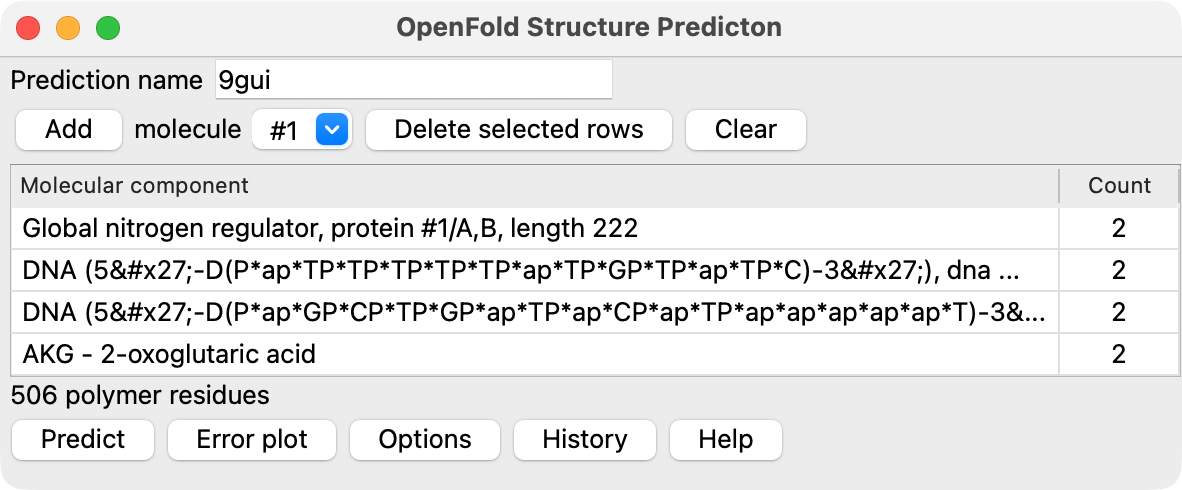
Predicting a structure with OpenFold
|
Menu of molecular components

|
To predict a structure made up of proteins, nucleic acids and small molecules you first specify
all the molecular components. Choose entries from the Add menu and press the Add button
to add them to your assembly specification in the table below. You can specify component molecules
in several ways.
- Protein and nucleic acids can be specified by choosing chains open models.
- Sequences of one letter codes for proteins (20 amino acids) or DNA or RNA can be pasted in.
- UniProt database identifiers can be give for proteins.
- Ligands, ions and solvent can be specified by 3-letter or 5-letter chemical component dictionary codes (e.g. ATP or HEM).
- Small molecules can be specified by SMILES strings.
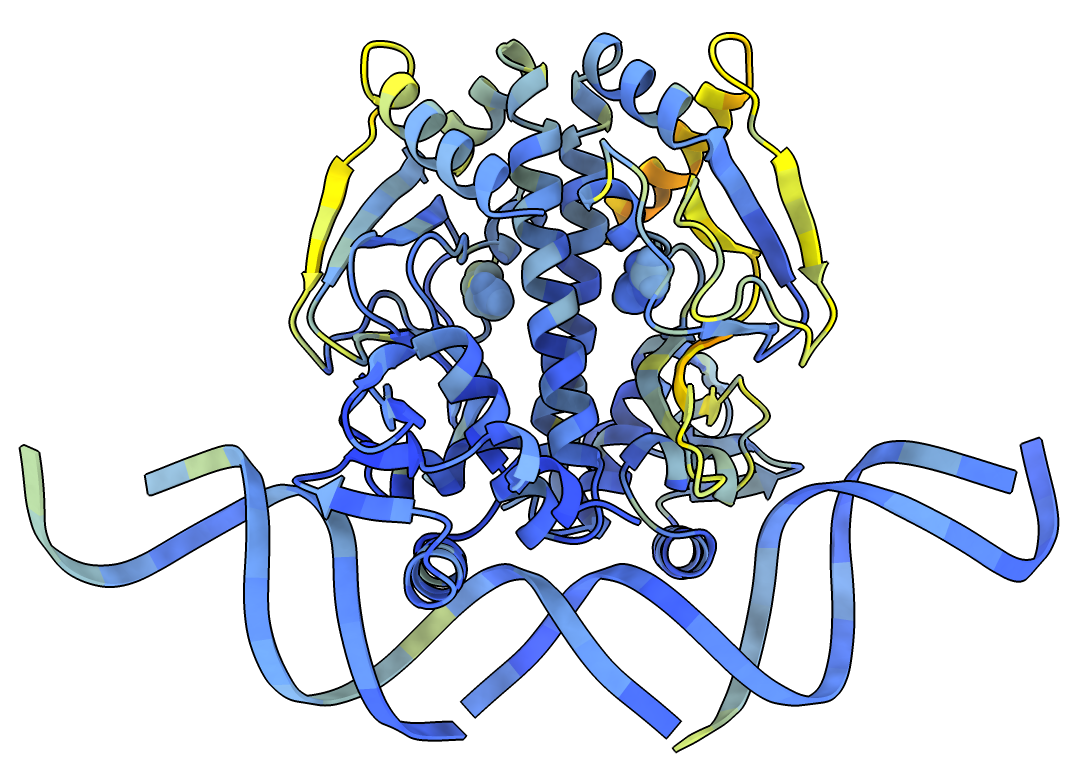
NTCA transcription factor bound to DNA
|
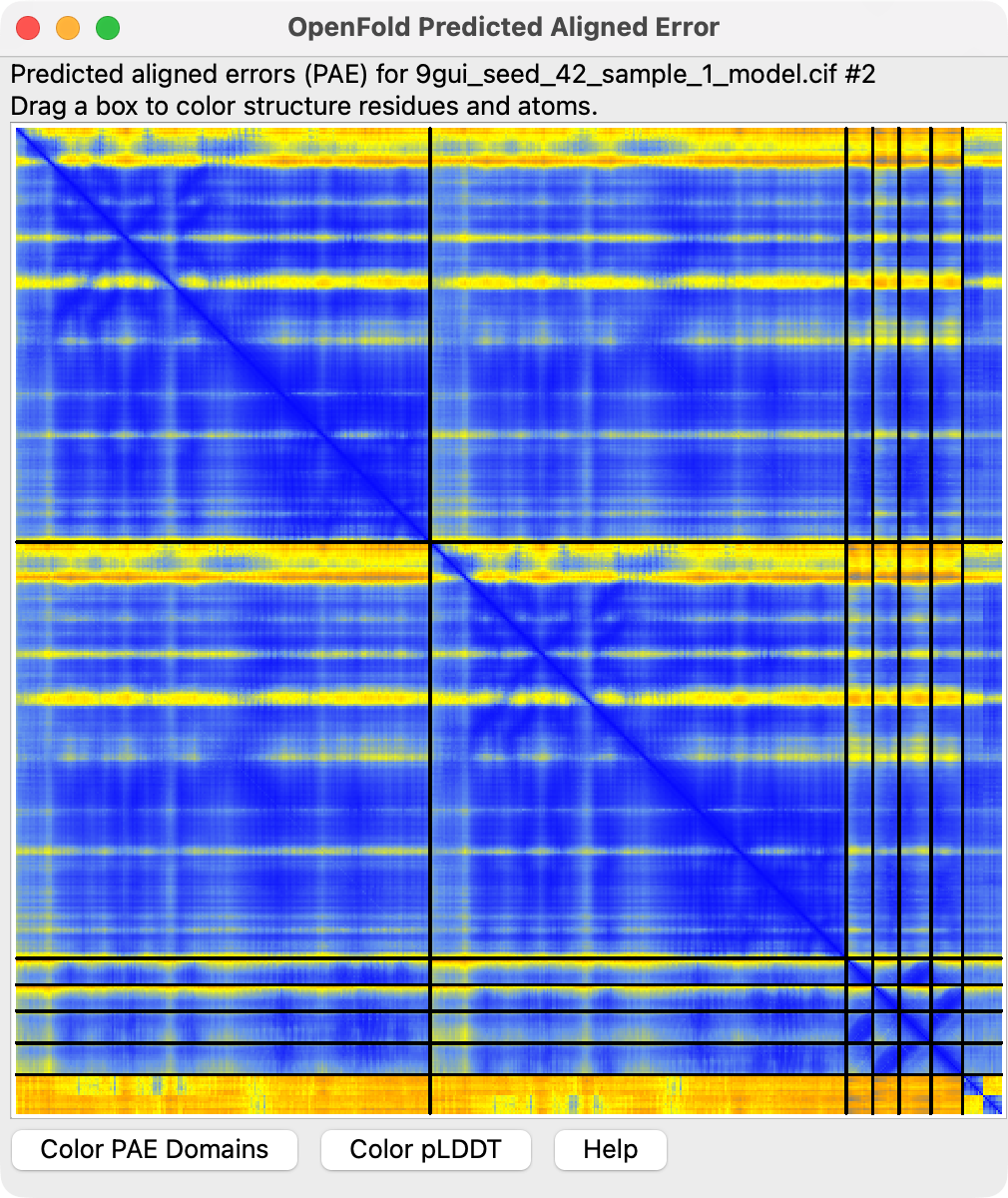
Predicted aligned error
|
Components can be added multiple times to have more instances of that molecule in the assembly.
Press the Predict button after the assembly is completed by adding each component to start the prediction.
A Stop button will be shown while the prediction runs to terminate the prediction,
discarding the partial computation so you can start another prediction.
Results
The results are put on your desktop in a new folder openfold/<prediction-name>
where the prediction name can be specified at the top of the ChimeraX OpenFold panel.
Using the Options described below you can change where the result files are placed.
Predictions for small assemblies, for example 500 residues and ligand atoms, take one to several minutes
depending on the computer (e.g. Nvidia GPU vs CPU only). Predictions run in the background (a separate process)
so you can continue to use ChimeraX while the calculation runs. The predicted structure will be opened
in ChimeraX when the calculation completes. If the assembly specification involved proteins or nucleic
acids specified using chains of open models, the predicted structure will be aligned
(using matchmaker)
to the open model for the first such component.
Coloring
The prediction will be colored using the standard AlphaFold pLDDT type of coloring where blue indicates
high confidence, yellow and red moderate to low confidence.
Predicted aligned error
Per-residue-pair estimates of prediction confidence can be displayed by pressing the Error Plot button
to show the predicted aligned error.
Structure size and prediction speed limitations
OpenFold structure predictions use a lot of memory and compute resources on your computer that limits the
size of the structure that can be predicted.
See the run times section below for example run times and size limits.
Mac.
It works well on Mac M-series (M1,M2,M3,M4) laptop and desktop
computers predicting small 100 residue structures in 1 minute and up to 1200 residues in about 15 minutes
with 32 GB of memory. With 16 GB of memory it can only predict about 350 amino acids taking about 5 minutes,
with larger predictions running out of memory.
Nvidia GPUs on Windows and Linux.
Nvidia GPUs on Windows and Linux computers also provide good performance.
On Linux with an Nvidia GPU with 24 GB of graphics memory (e.g. Nvidia RTX 3090 or 4090)
it can predict about 1300 residues in about 4 minutes, larger sizes run out of memory.
Testing on Windows with less GPU memory, 12 GB (e.g. Nvidia RTX 4070) predicts up to 1000 residues.
On Windows with 8 GB of GPU memory (e.g Nvidia RTX 3070) predictions of about 700 residues.
Large predictions on Nvidia GPUs on Linux run out of memory and fail. On Windows the prediction
will fallback to using CPU memory allowing larger structure predictions but taking immensely longer
run times (10-30 times longer).
Intel CPU.
Predictions only utilizing an Intel CPU are very slow, for example 1.5 hours for 900 residues.
Expected size limits are about 350 residues with 16 GB, 1000 residues with 32 GB, and about 1600 residues
with 64 GB. Run time is expected to increase as the square of the number of residues.
Run times and size limits
Here are run times for a few desktop and laptop computers for predicting various size molecular assemblies
from the Protein Databank using the ChimeraX OpenFold 3 fork
from February 11, 2026.
OpenFold prediction times in minutes, using cached MSAs. Tokens is the number of polymer residues plus ligand atoms.
| PDB code
| Tokens
| Mac
M1
16 GB
| Mac
M1 Max
32 GB
| Mac
M2 Ultra
64 GB
| Linux
i9 CPU 64 GB
| Linux
Nvidia 4090
| Windows
i7 CPU 64 GB
| Windows
Nvidia 3070
| Number of residues and atoms and prediction error
|
| 8rf4
| 129
| 1.5
| 1.0
| 0.8
| 1.4
| 0.7
|
| 1.5
| 118 amino acids, 11 ligand atoms, 1.5A RMSD 118 residues
|
| 9gui
| 526
| 18
| 4.3
| 2.5
| 16
| 1.0
|
|
| Protein dimer, DNA, 2 ligands, 1.4A RMSD for protein
|
| 9moj
| 660
| 30
| 6.6
| 3.7
| 25
| 1.1
| 30
| 3.3
| 660 amino acids, heterotetramer, 0.9A RMSD 125 residues
|
| 9h1k
| 671
|
| 6.9
| 4.0
|
| 1.1
|
|
| 560 amino acids, 59 rna bases, 52 ligand atoms,
1.2A RMSD for 261 residues, RNA wrong
|
| 9b3h
| 911
|
| 38
| 7.6
|
| 1.5
|
|
| 911 amino acids, heterodimer, 1A RMSD 503 residues
|
| 9fz5
| 1025
|
|
| 11
|
| 1.7
|
|
| 1025 amino acids, heterotrimer, 2.2A RMSD 740 residues
|
| 9mcw
| 1154
|
|
| 16
|
| 2.3
|
|
| 1154 rna bases, homodimer, wrong dimer and monomer conformations
|
| 8sa0
| 1371
|
|
| 30
|
| 2.6
|
|
| 1274 amino acids, 97 ligand atoms, 2.1A RMSD 1151 residues
|
| 9gh4
| 1467
|
|
|
|
| 2.9
|
|
| Protein homotrimer, monomer 489 residues, 1.5A RMSD for 250 residues
|
| 9enr
| 1794
|
|
|
|
| 4.1
|
|
| Protein monomer, 4 ligands, 2.9A RMSD for 1551 residues
|
| 1dpp
| 2028
|
|
|
|
| 5.2
|
|
| Homotetramer, 0.47A RMSD for 507 residues
|
| 9hma
| 2218
|
|
|
|
| failed
|
|
| Protein monomer
|
- Mac M1 16 GB - Mac Mini, 8 core GPU, euclid.cgl.ucsf.edu
- Mac M1 Max 32 GB - MacBook Pro 32 core GPU, Tom's laptop
- Mac M2 Ultra 64 GB - Mac Studio 60 core GPU, descartes.cgl.ucsf.edu
- Linux i9 CPU - cpu i9-13900K (24 cores) 64 GB (DDR5 5200MHz), minsky.cgl.ucsf.edu
- Linux Nvidia 4090 - VRAM 24 GB, cpu i9-13900K (24 cores) 64 GB (DDR5 5200MHz), minsky.cgl.ucsf.edu
- Windows i7 CPU - cpu i7-12700K (12 cores) 64 GB DDR5 4000MHz, vizvault.cgl.ucsf.edu
- Windows Nvidia 3070 - Windows 11, VRAM 8 GB, PCIe 4.0, cpu i7-12700K (12 cores) 64 GB DDR5 4000MHz, vizvault.cgl.ucsf.edu
Performance notes
Mac GPU acceleration.
The reported Mac performance is for Mac M1/M2/M3/M4 series GPUs.
OpenFold uses machine learning package torch which
has GPU acceleration called Metal Performance Shaders (MPS) on these Mac M series GPUs
which have speed up to 2-5x slower than an Nvidia 4090 but with the advantage that the
Mac can handle larger molecular systems using the unified computer memory (e.g. 32 or 64 GB).
With 16 GB prediction size is limited to 350 residues.
Older Mac Intel machines do not have GPU acceleration in Torch
and run at speeds similar to Windows Intel CPU-only predictions.
Windows Nvidia GPU performance.
The above table shows a significant slow-down in predictions beyond about 600 residues on Windows with Nvidia 3070 (8 GB) and 4070 (12 GB) graphics. This is probably because the GPU memory is insufficient for larger structures and the machine learning toolkit falls back to a mix of CPU and GPU calculation. Notice that the 4070 GPU took more time than the 3070 GPU for large structures probably because the CPU on the 4070 machine (i5-6700K) is significantly slower than the CPU on the 3070 machine (i7-12900K).
Linux Nvidia GPU out of memory.
On Linux Nvidia 4090 with 24 GB of GPU memory the maximum prediction appears to be about 2000 residues plus ligand atoms before an "out of memory" error occurs. This contrasts with Windows where Torch appears to fallback to using CPU and not run out of GPU memory.
CUDA use of bfloat16.
On Nvidia CUDA the ChimeraX OpenFold uses 16-bit floating point (bfloat16, bf16-mixed torch lightning) which
allows larger predictions then on non-CUDA systems where 32-bit float is used. Torch only supports hardware
accelerate bfloat16 CUDA .
Batch predictions with sets of ligands
Several predictions can be run each with a different ligand to see how it binds to the rest of the molecular assembly. Use the Add molecule "each ligand SMILES string" menu entry. Paste in a set of SMILES strings with ligand names. Each line should have a ligand name, comma, followed by a SMILES string. The ligand names will be used as the predicted structure file names. Alternatively you can just enter one SMILES string per line without a name and the ligands will be named "ligand1", "ligand2", .... Press the Add button after pasting in the set of ligands, then the Predict button to run the series of predictions.
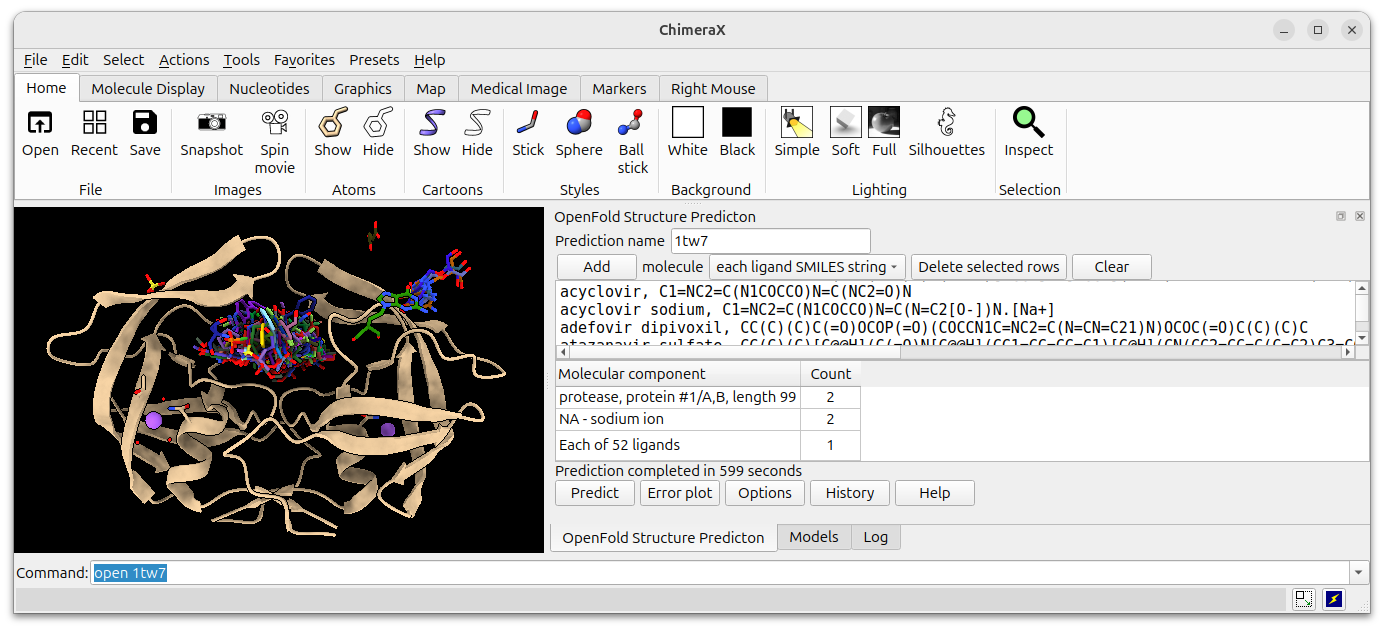
Example input for 52 anti-viral ligands run against HIV protease.
Predictions completed in 10 minutes on an Nvidia 4090 GPU.
| 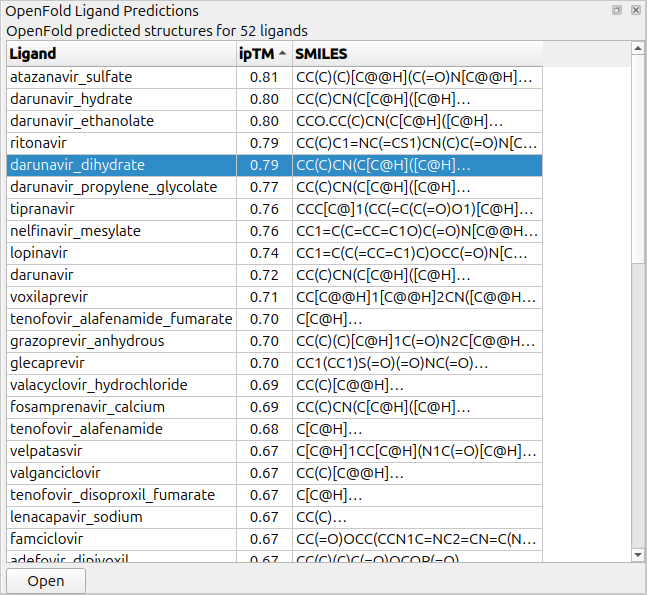
Table of prediction ipTM confidence for ligands.
|
Progress messages will appear in the OpenFold panel as the structures are predicted for each ligand.
The ipTM scores is a crude prediction of binding confidence.
When the predictions complete a table of results giving
ligand name, ipTM and SMILES string will appear. The table can be
sorted by clicking on the column headers. Selecting rows of the table and pressing the Open button
will open the selected structure predictions. The table is written as comma-separated values to the
directory where OpenFold was run, for example hiv_protease/hiv_protease.oflig in the case shown.
This file will appear in the file history thumbnails when you start ChimeraX in future sessions
and clicking on the thumbnail will reshow the table.
Options
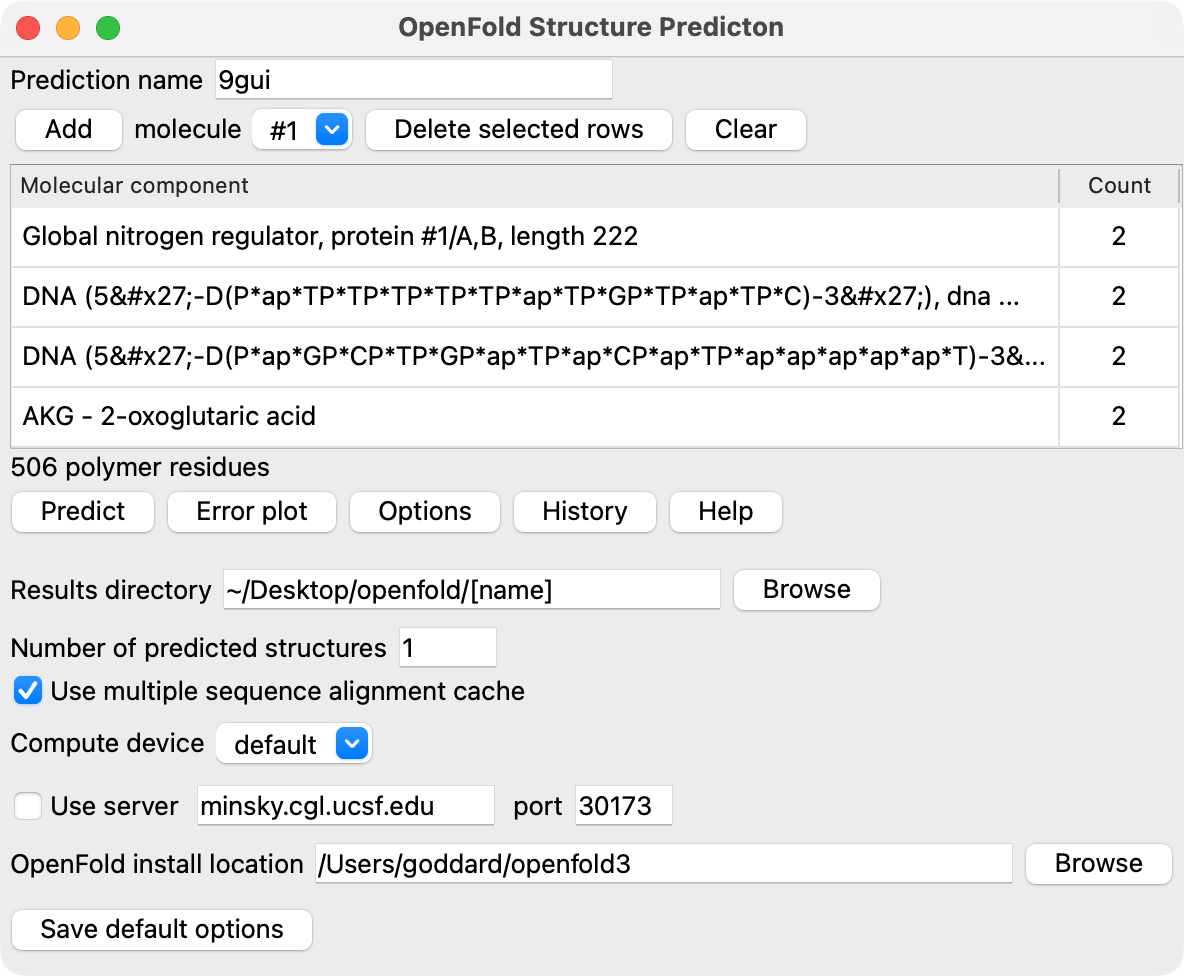
Pressing the Options button shows additional settings for openfold predictions.
- Results directory: You can change where the results are placed.
The path contains "[name]" which is replaced with the prediction name.
- Number of predictions: The default number of predicted structures is 1, but you can request more to get an ensemble of structures that usually have small variations.
- Use multiple sequence alignment cache: OpenFold uses deep multiple sequence alignments for proteins
that are computed using the Colabfold server. To avoid recomputing the alignments when you run predictions
with the same proteins but different ligands ChimeraX caches those alignments in ~/Downloads/ChimeraX/OpenFoldMSA
and the cached alignments will be used if available as long as this option enables using the cache.
- Compute device: The computation can run on Nvidia GPUs or Mac M series GPUs and complete faster than CPU-only calculations. The default setting tries to uses the GPU if available.
- OpenFold install location: OpenFold is installed in a virtual Python environment contained in this folder.
- Save default options: Save the currently shown option settings as the defaults for future ChimeraX sessions.
Additional advanced options are available by using the ChimeraX openfold command.
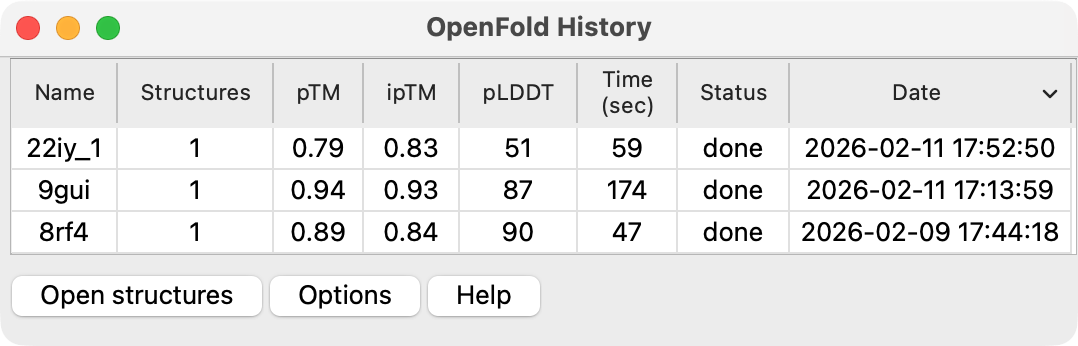
Listing past predictions
ChimeraX menu entry Tools / Structure Prediction / OpenFold History lists a table of past
OpenFold predictions. Selecting rows of the table and pressing the Open structures button
will open those structures. The table is filled by scanning the OpenFold prediction directories
located in the directory where ChimeraX puts new OpenFold predictions (by default ~/Desktop/openfold).
If a prediction computed multiple structures they will all be aligned to the first structure
of that prediction when opened (using the ChimeraX matchmaker command). The directory containing the prediction
directories and the choice to align can be specified in the panel shown by pressing the Options button.
Directories are identified as containing OpenFold predictions if they contain a file named "command".
When ChimeraX runs a prediction it saves the openfold command including all its arguments are listed
in this file.
Fetching server predictions
If there are server predictions that did not complete before
quitting ChimeraX, then when ChimeraX is run again the OpenFold History panel will show a button
Fetch from server next to the Open structures button. This button fetches any server
predictions that have completed and opens them. Predictions that have
not yet completed are noted in the Log after pressing this button.
ChimeraX openfold command
The ChimeraX OpenFold graphical interface runs a prediction by running the ChimeraX openfold command.
That command is recorded in the ChimeraX Log panel, and looking at that command can help you understand
the command options.
openfold predict [sequences] [protein sequences] [dna sequences] [rna sequences]
[ligands residue-spec] [excludeLigands ccd-codes]
[ligandCcd ccd-codes] [ligandSmiles smiles-string]
[forEachSmilesLigand name,smiles-string,name,smiles-string...]
[name prediction-name] [resultsDirectory directory] [samples n] [seeds s1,s2,...]
[device default|cpu|gpu] [precision 32-true | bf16-mixed | 16-true | bf16-true]
[useServer true | false] [serverHost hostname] [serverPort port]
[useMsaCache true|false] [msaOnly true|false]
[open true|false] [installLocation directory] [wait true|false]
Options descriptions
- sequences - Sequences can be specified using chain ids, UniProt identifiers, explicit strings of amino acid 1-letter codes, or ChimeraX sequence viewer ids.
- protein sequences - Like the previous sequences option only this explicitly excludes non-protein sequences in specifiers for open models. For example "protein #1" would not include any DNA/RNA chains of model #1. This option can be used more than once.
- dna sequences - Uses only DNA sequences from open model specifiers and treats explicit 1-letter code sequences as DNA. UniProt ids cannot be used. This option can be used more than once.
- rna sequences - Uses only RNA sequences from open model specifiers and treats explicit 1-letter code sequences as RNA. UniProt ids cannot be used. This option can be used more than once.
- ligands residue-spec - Specify ligands using residue specifiers for open models.
- excludeLigands ccd-codes - Exclude these CCD codes when interpreting the ligands option. By default it excludes ccd code "HOH", ie water.
- ligandCcd ccd-codes - Comma-separated list of 3 or 5-letter CCD codes. This option can be used more than once.
- ligandSmiles smiles-string - Comma-separated list of SMILES strings. This option can be used more than once.
- forEachSmilesLigand name,smiles-string,name,smiles-string... - Comma-separated list of ligand names and SMILES strings. A separate prediction will be made for each ligand by adding each ligand to the molecular assembly.
- name prediction-name - The name of the prediction directory that will be created.
If the folder already exists an numeric suffix _1, _2, _3... will be appended.
- resultsDirectory directory - Path to the results directory that will be created. "[name]" in the path will be replaced by the prediction name. If the folder already exists an numeric suffix _1, _2, _3... will be appended.
- samples n - Number of predictions. Default is 1. This is what OpenFold calls "diffusion samples". Creating additional structures takes much less time than creating the first structure.
- seeds n - Comma-separated list of random number seeds (integer) to initialize calculation. A separate prediction is run for each seed. Runs with different seeds will give different results. Default seed is 42.
- device default | cpu | gpu - Whether to run the computation on GPU or CPU. The default setting chooses based on GPU availability and torch support for the GPU.
- precision 32-true | bf16-mixed | 16-true | bf16-true - Whether to use 32-bit floating point or 16-bit floating point precision in predicting structures. These are the Torch Lightning names for different precisions. 32-true uses float32. The other modes all use 16-bit floating point with 16-true using float16 which uses 11-bit mantissa and 5-bit exponent giving limited range (+/-65504), or bf16-true using bfloat16 with 8-bit mantissa and 8-bit exponent giving the same range as float32 but with reduced precision. The bf16-mixed mode uses bfloat16 for most operations and float32 for others that need higher precisions. Default is bf16-mixed for predictions using Nvidia GPUs with CUDA, and 32-bit on other platforms. Using 16-bit modes allows running larger structure predictions without running out of memory but those modes are only currently supported by Torch in hardware when using CUDA.
- useServer true | false - Whether to run computations on a prediction server specified by the serverHost and serverPort options.
- serverHost hostname - The host name of the server, for instance minsky.cgl.ucsf.edu or 169.230.21.20. The default uses the python gethostbyname() system call to get the host name or IP address.
- serverPort port-number - The port number to listen for connections. Default 30172.
- useMsaCache true | false - Whether to use protein deep sequences alignments from the ChimeraX OpenFold MSA cache in ~/Downloads/ChimeraX/OpenFoldMSA. Because the alignments for different proteins in an assembly are paired to match ones from the same organisms, using the cache requires that an assembly have the exact same set of proteins. It cannot use alignments computed for individual proteins from multiple different runs.
- msaOnly true|false - Whether to just calculate and cache the multiple sequence alignment using the Colabfold server. This allows computing the MSA separately from the structure. It can be useful to precompute and cache MSAs when running structure predictions on a cluster which does not have internet access.
- open true | false - Whether to open the predicted structures when the prediction finishes.
The structures will be aligned to an already open model if that open model was used (the first used) in
specifying the assembly.
- installLocation directory - Where OpenFold is installed. If specified this sets the default location used in future ChimeraX sessions.
- wait true | false - Whether ChimeraX should wait frozen while the prediction is computed or return immediately and allow ChimeraX use during the computation.
Installation command
openfold install [directory] [downloadModelWeights true | false] branch name
The openfold install command creates a Python virtual environment to install the
ChimeraX OpenFold fork Github. If no directory
is specified then ~/openfold3 in the user's home directory is used. The directory
will be created or if it already exists must be empty. It then downloads
the OpenFold network parameters to ~/.openfold3.
The install uses a fork of the OpenFold repository https://github.com/RBVI/openfold-3. It uses git branch chimerax_openfold of this fork unless the branch option
is specified in which case it installs the specified branch.
The install process executes these commands to make the virtual environment and install OpenFold.
It uses the ChimeraX Python executable to create the virtual environment. OpenFold will no longer
work if ChimeraX is moved or deleted and will need to be reinstalled in that case. It will
also no longer work if the openfold directory itself is moved since the openfold executable
refers to the install location to find python.
The ChimeraX openfold install command creates a Python virtual environment and installs
openfold and downloads the openfold weights.
On Windows it installs a version of torch with CUDA 12.6 support before installing
openfold if Nvidia graphics is detected.
python -m venv directory
directory/bin/python -m pip install torch --index-url https://download.pytorch.org/whl/cu126 # On Windows with Nvidia GPU only.
directory/bin/python -m pip install openfold
directory/bin/python chimerax/site-packages/openfold/download_weights.py
Ligand table command
openfold ligandtable runDirectory [ alignTo atomic-model ]
The openfold ligandtable command displays a table for batch ligand predictions.
Normally this command is not needed because ChimeraX batch ligand predictions are tabulated in
the directory where the OpenFold run is made in a comma-separated value file with suffix ".oflig"
and you can open this file to show the table of results. But in cases where this file is missing
you can recreate the table with this command which searches the prediction results extracting
the scores, and searches the ".json" input file to find the SMILES strings for the ligands.
Limitations
- Structure size. OpenFold uses a lot of memory and the amount of available memory
limits the size of structures that can be predicted. For a computer with 32 Gbytes the size limit
is roughly 1000 residues plus ligand atoms (called "tokens"). Consumer Nvidia GPUs with 8 or 12 GB of
memory (e.g. RTX 3070) only handle 300-500 residues before using CPU memory on Windows that slows
the prediction speed by 10-20 fold. On Linux it will not use CPU memory. Consumer Nvidia GPUs with
24 GB (RTX 3090 and RTX 4090) are able to predict 1300 tokens.
Prediction size limits are perhaps the most important
shortcoming of OpenFold compared to AlphaFold 3 which handles memory more efficiently and is able to
predict 5000 tokens with 80GB of GPU memory, about twice the size that OpenFold can predict.
A drawback of AlphaFold 3 is that it requires Linux and an Nvidia GPU in addition to various
licensing restrictions. We hope in the future OpenFold will optimize memory use to predict larger
structures.
- Run time. The computation time increases as the square with the number of tokens. So
a prediction with 3 times the number of residue and ligand atoms will take approximately
9 times longer to run.
- Nvidia GPU support on Windows.
Installing OpenFold will get a CUDA-enabled version of the torch machine learning package
if it detects Nvidia graphics. It decides if you have Nvidia graphics by seeing if the file
C:/Windows/System32/nvidia-smi.exe exists. Otherwise it gets a cpu-only version of torch.
If you install an Nvidia graphics driver after installing OpenFold you will have to reinstall
OpenFold to get the CUDA version. The installed torch is for CUDA 12.6 or newer.
If your computer has a version of CUDA older than 12.6 but newer than 11.8 you can run the
following commands in a Windows Command Prompt to install a CUDA 11.8 version of torch.
For other CUDA versions refer to the
Torch installation page
for the correct pip install command.
> cd C:\Users\username\openfold\Scripts
> pip.exe uninstall torch
> pip.exe install torch --index-url https://download.pytorch.org/whl/cu118
- Nvidia GPU support on Linux.
On Linux the installed OpenFold will work with CUDA 12.6 or newer if you have Nvidia graphics.
If you have an older system CUDA version it may still work, or you can refer to the
Torch installation page
for the correct pip install command and replace torch with the following shell commands.
$ cd ~/openfold3/bin
$ ./pip uninstall torch
$ ./pip install torch --index-url https://download.pytorch.org/whl/cu118
- No assigning chain identifiers. It can be helpful to assign chain identifiers (A,B,C...) to
the different molecular components to match existing structures. OpenFold is capable of this but the
ChimeraX user interface does not currently allow it.
- MSA sequence alignments. OpenFold uses the Colabfold MSA server (https://api.colabfold.com)
for computing deep sequence alignments. This requires internet connectivity and is subject to
outages if that server is down. The sequence alignments are cached
in ~/Downloads/ChimeraX/OpenFoldMSA so subsequent predictions with the same set of protein sequences
can reuse the sequence alignment.
Change log
- February 13, 2026. Initial OpenFold prediction tool using OpenFold 3 preview 1 from January 2026.
- March 25, 2026. Updated ChimeraX to use OpenFold preview 2 using github primary code from March 24, 2026.
OpenFold modifications
ChimeraX uses a fork of the OpenFold repository
with several improvements summarized below. The exact changes are seen in
openfold_diffs.patch which compares the March 25, 2026 ChimeraX OpenFold
to the primary OpenFold repository main branch
using command git diff unmodified chimerax_openfold_preview2.
- Compute and write PAE.
By default OpenFold computed predicted aligned error (PAE) but did not have any option to write it
to a file. I added it to the confidence output since it is the most heavily used metric for detailed
analysis of predictions. Also I changed the default confidence file output from json format (.json)
to numpy format (.npz) which is many times smaller and many times faster to read.
- Don't remove MSA and template files.
OpenFold puts MSA and template files in a temporary directory and deletes them.
These files are valuable for understanding the confidence of the predicted structures
so ChimeraX writes them to the prediction output directory and does not delete them using
standard OpenFold settings. But the OpenFold code always deletes the raw MSA (.a3m file)
results from Colabfold and I changed it to preserve those as well.
- MSA only option.
Sometimes it is useful to only compute MSAs in one run, and then predict structures in another run.
For instance if the same MSA will be used for hundreds of protein complexes with same same proteins
but different drugs. Also it is needed for runs on compute clusters that don't allow internet, where
the MSA calculation needs to be run on one computer, and the inference on another. I added a
--msa_and_templates_only OpenFold command-line option to handle this.
- Seeds option.
I added a --seeds option that takes a comma-separated list of integers. A separate prediction
is run for each seed. Different seeds will produce different predictions which may be more diverse
because the PairFormer network is rerun, while predicting multiple samples only reruns the atom
diffusion stage. Default seed is 42.
- Compute device option.
I added a --device option (values "gpu", "cpu", "tpu") to control what device a prediction uses.
The default is GPU, but for consumer GPUs with little memory (e.g. 8 or 12 GB) larger structure
predictions will run out of memory and being able to run the computation more slowly on CPU allows
it to complete successfully.
- Precision option.
This specifies floating point precision to use bf16-mixed or 32-true. Standard OpenFold always
uses 32-bit precision which uses twice the memory as 16-bit precision. Boltz and other prediction
software saw no decrease in accuracy with 16-bit precision for inference. In the torch current
machine learning package only CUDA has hardware supported 16-bit. In ChimeraX OpenFold it uses
bf16-mixed which is a Torch Lightning precision that uses bfloat16 for most operations but 32-bit
for some that are known to require higher precision. This allows running significantly large
predictions on Nvidia GPUs, for example, 2000 residues vs 1300 residues max size on an Nvidia RTX 4090
with 24 GB of memory. By default ChimeraX OpenFold uses bf16-mixed on CUDA (Linux or Windows) and
32-float on Mac GPU or Windows CPU. I added a --precision option to allow specifying the precision
if testing for future Torch versions or for accuracy degradation is needed.
- Download OpenFold parameters without user input.
OpenFold installation requires the user to some choices about whether and where to download
OpenFold model parameters. I modified the setup_openfold.py script so it can to the setup
non-interactively. ChimeraX installs OpenFold non-interactively.
- Operating system specific packages.
Several packages used by OpenFold are not available on Mac or Windows and are also not required
but they cause the standard OpenFold install to fail. I modified the pyproject.toml dependency file
to not attempt to install from PyPi mkl, aria, nvidia-cutlass, cuda-python on Mac since they are
not available. Also I made it not require deepspeed or kalign-python on Windows. Extra code was
added to use a kalign executable on Windows (and Mac). The other packages are for optimization or
downloads and are not necessary for predictions.
- Include kalign binaries. In order for ChimeraX to install the kalign sequence alignment program
used by OpenFold to align template structures to query sequences I put kalign executables for Mac, Windows,
and Linux in the ChimeraX OpenFold repository under kalign. The standard OpenFold uses conda to install kalign which is only available for Linux and Mac.
OpenFold preview 2 switched to using the kalign-python PyPi package. ChimeraX uses that on Linux but
uses the kalign executables on Windows because the PyPi packages is not available for Windows, and also
on Mac because the kalign-python package fails on Mac due to duplicate libomp.dylib libraries (one included in the kalign package and another in OpenFold).
- DeepSpeed only on Linux.
OpenFold uses DeepSpeed to accelerate CUDA GPU computations and has it enabled by default.
DeepSpeed is not available on Mac or Windows, so I make the default setting not use it on those operating
systems. Also DeepSpeed is disable for Linux CPU (without CUDA) predictions.
- Mac multiprocessing fix.
OpenFold uses the multiprocessing module with pickling to call an data processing function.
On Linux this works with the function defined within another function, but on Mac the function
must be at global scope. I moved the function to global scope.
- Remove poor performance multi-processing.
OpenFold uses multi-processing presumably to speed-up processing training batches (num_workers = 10,
num_workers_validation = 4 in validator.py). But this slows down inference where the batch size is
one query and spinning up the subprocesses takes several seconds. I changed it to not use multiprocessing
making predictions several seconds faster.
- Remove developer debugging log messages.
OpenFold outputs code debugging messages in several places using logger.info() or print()
instead of logger.debug() producing clutter in the output meaningful only to developers.
I changed those to logger.debug() which is not output unless debug mode is enabled.
Makes it easier to see real problems in the log.
- Progress messages.
To allow ChimeraX to show sensible progress messages as a prediction is made I added log output
that says when each stage that takes significant time starts and stops (e.g. executable startup,
loading weights, colabfold search, template processing, feature processing, inference).
- Turn off progress bars.
The tdqm progress bars create hundreds of output in log files
and are only useful for output to at terminal. Turn those off by default.
- Log time stamps.
To identify why some predictions take a long time it is useful to include time stamps on log output
lines. For instance this allows ChimeraX to report that OpenFold took several minutes waiting for
a Colabfold MSA calculation to complete. Also it helps identify bottlenecks (e.g. most of the time of
for predicting small structures is not in inference).