
from Guerrero-Ferreira R, eLife 2018.
|
|
| 2D electron micrograph of alpha-synuclein fibrils from Guerrero-Ferreira R, eLife 2018. |
Tom Goddard
BCBB Training Seminars
Sept 11, 2019
We look at basic ChimeraX capabilities for display and analysis of atomic models using recent alpha-synuclein fibril structures from cryoEM. Using ChimeraX version 0.91 daily build. User's Guide and other tutorials.
Alpha-synuclein is a small protein of 140 amino acids that aggregates in neuronal cells in Parkinson's disease. It is believed to form fibrils in intracellular compartments called Lewy bodies. At least 4 very different fibril atomic models have been determined by cryoEM in the last few years, all from recombinant preparations. Models from fibrils exctracted from brain tissue have not yet been determined. Six mutations (A30P, E46K, H50Q, G51D, A53E, and A53T) are associated with Parkinson's disease or alpha-synuclein aggregation, although these account for extremely few cases of disease. A truncated version of alpha-synuclein leaving residues 1-121 is much more prone to aggregation and is used in the cryoEM studies.
We will look at structures (PDB 6h6b, EMDB 0148 and 4276) described in
Cryo-EM structure of alpha-synuclein fibrils.
Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H.
Elife. 2018 Jul 3;7. pii: e36402. doi: 10.7554/eLife.36402.
and (PDB 6rt0, EMDB 4994 and 4996)
bioRxiv preprint, May 2019
Two new polymorphic structures of alpha-synuclein solved by cryo-electron microscopy
Ricardo Guerrero-Ferreira, Nicholas M.I. Taylor, Ana-Andrea Arteni, Pratibha Kumari, Daniel Mona,
Philippe Ringler, Markus Britschgi, Matthias E. Lauer, Joeri Verasdock, Roland Riek, Ronald Melki,
Beat H. Meier, Anja Böckmann, Luc Bousset, Henning Stahlberg
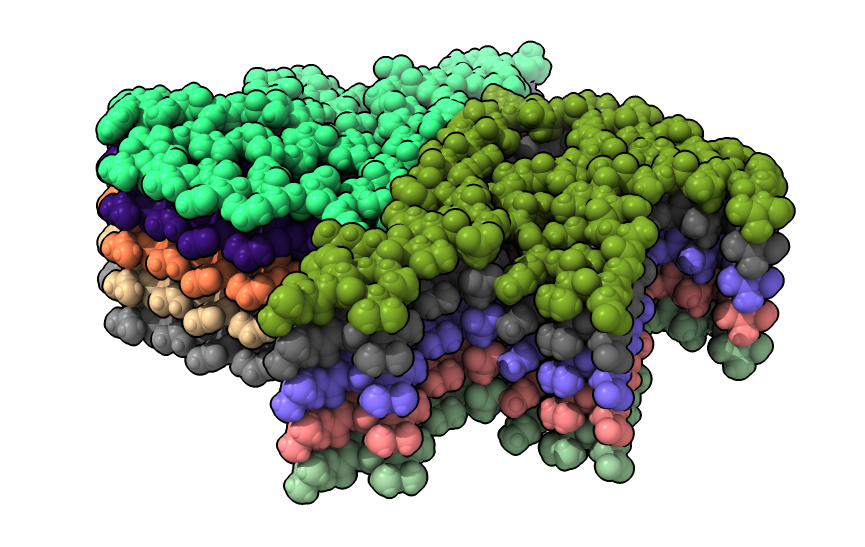
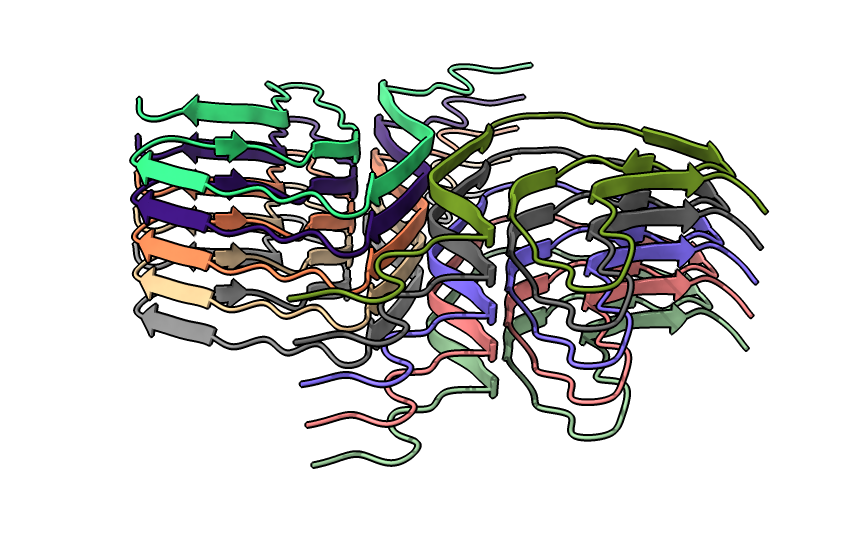
| 
| 
|
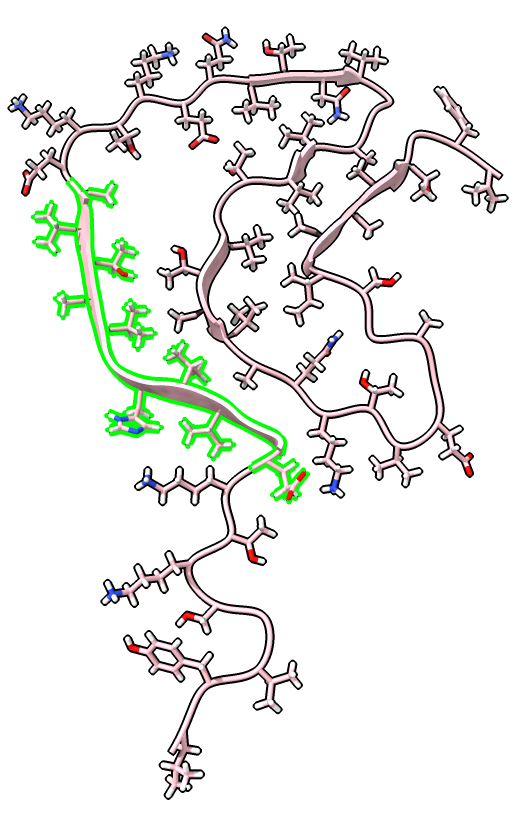
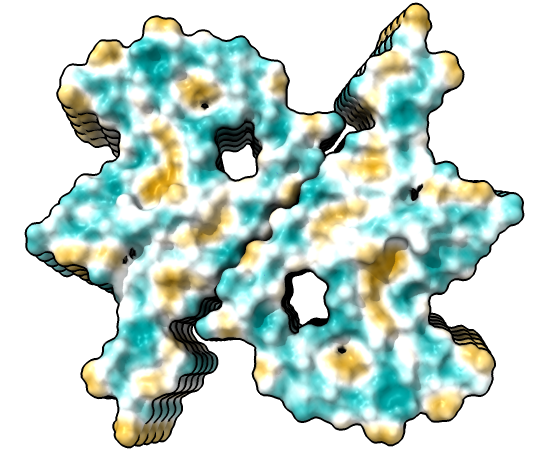
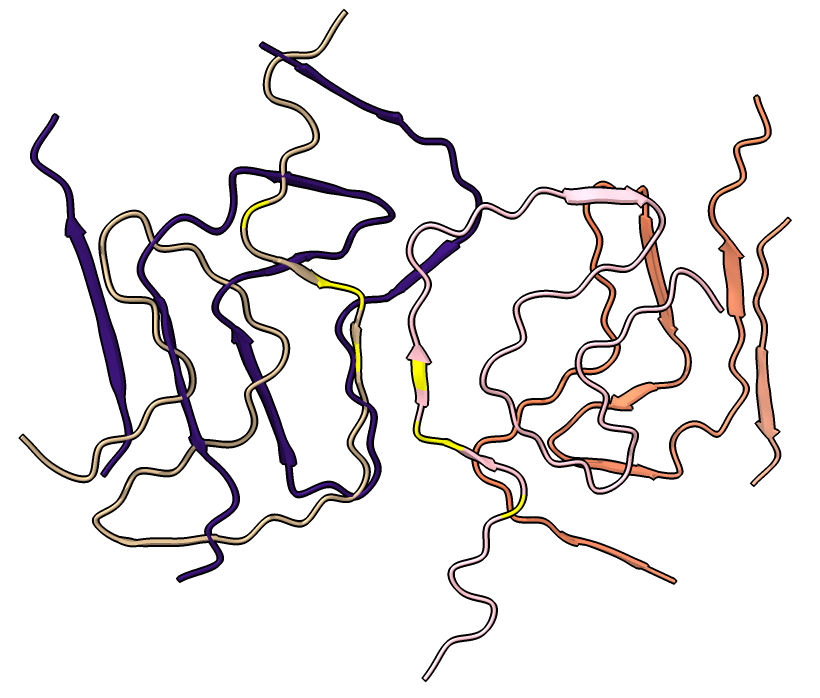
| open 6h6b | hide atoms show ribbons | Looking down fibril axis. |

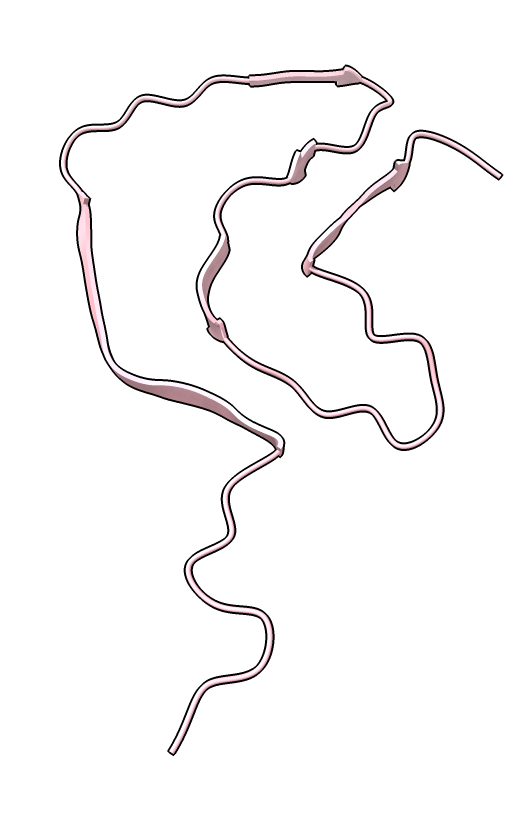
| 
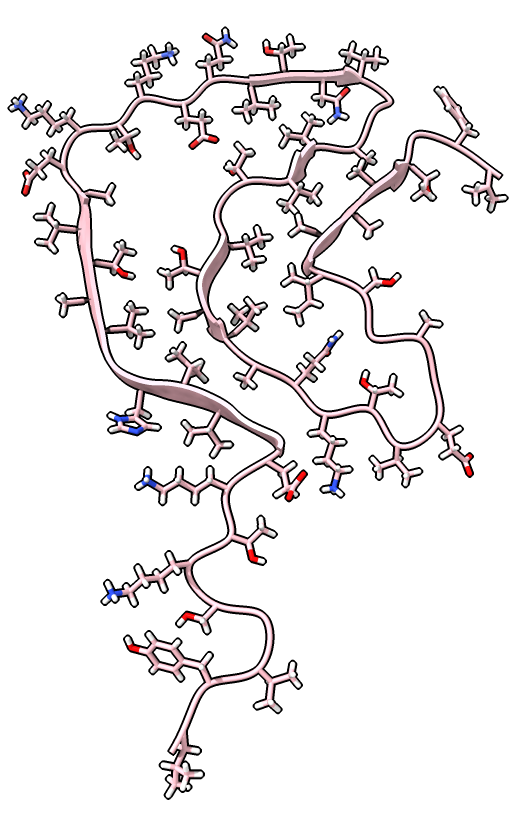
| 
|
| color /D,E,F,I,J tan color /A,B,C,G,H pink | show /A ribbon only | show /A atoms style stick color byhet |
Details

| 
|
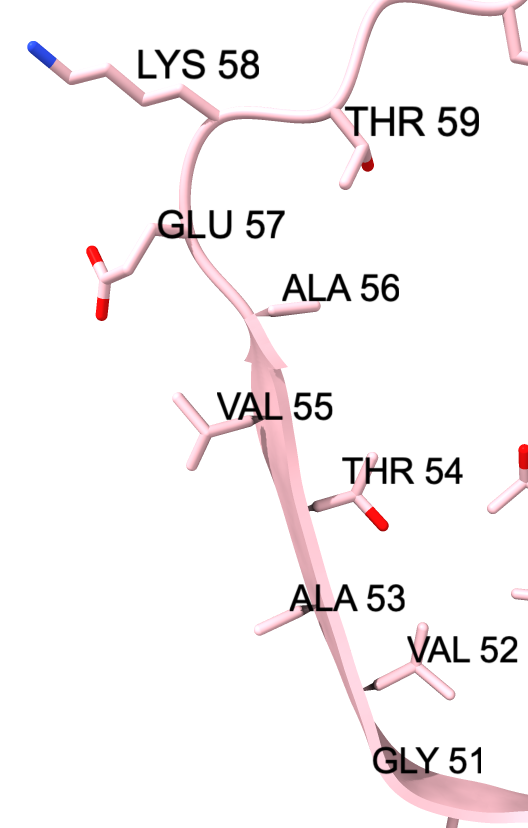
| Chain table from log. | Click alpha-synuclein link to show amino acid sequence. |
| 
| 
|
| Ctrl-click ribbon to select residue.
Up-arrow key to select beta strand. Ctrl-click background to clear selection. | Selection also shows on sequence view. | label /a label height 1.5 hide H label delete |
Details
| 
| 
|
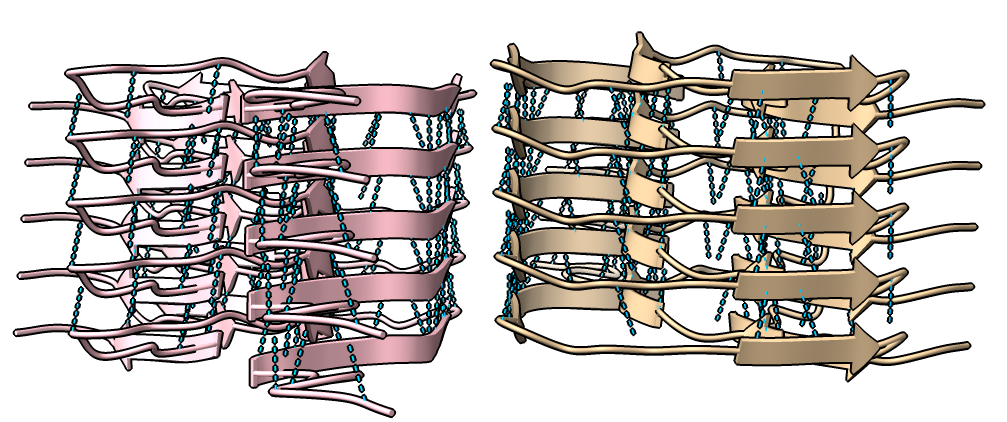
| show ribbons hide atoms hbonds ~hbonds | show surfaces or use Molecule Display toolbar | mlp or use Molecule Display toolbar |
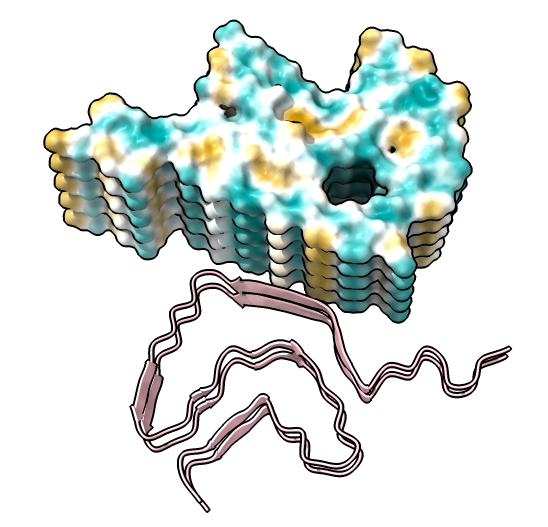
| 
|
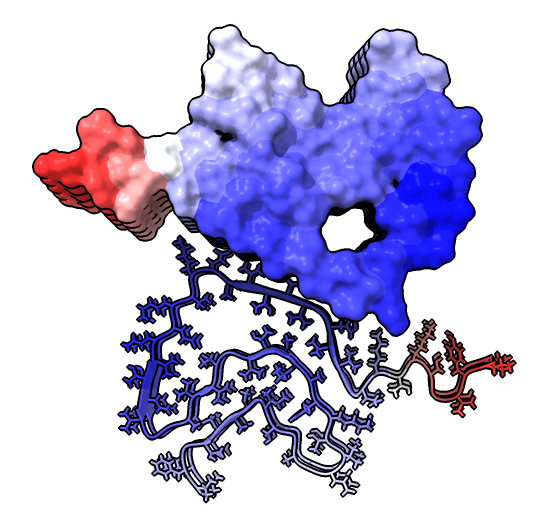
| hide /A,B,C,G,H surface hide /A,B,C ribbon Dimer interface is not very hydrophobic. | color bfactor show /G,H atoms Blue is highest resolution, red lowest. |
Details
|
Rotation angle (degrees) 179.51228273 Shift along axis 2.44997643 RMSD between 806 atom pairs is 0.007 angstroms | 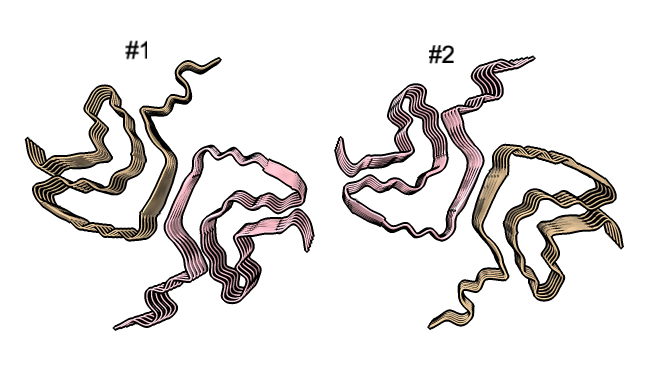
|
|
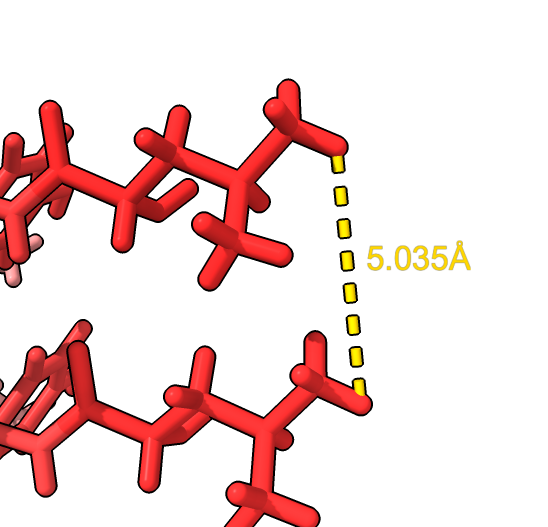
Use distance mouse mode from toolbar and click two atoms
distance /H:38@HD12 /G:38@HD12 surface close ; hide ribbon ~distance | open 6h6b
align #2/G toAtoms #1/I reportMatrix true | tile close #2 |
Details
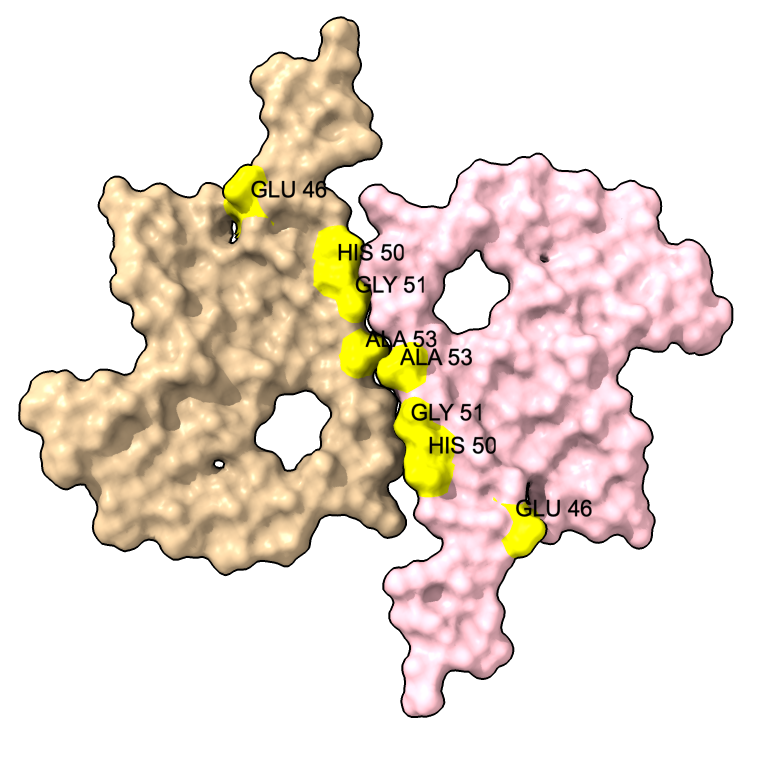
We can show some the location of disease associated mutations A30P, E46K, H50Q, G51D, A53E, and A53T (from Guerrero-Ferreira, eLife 2018).
| 
| 
|
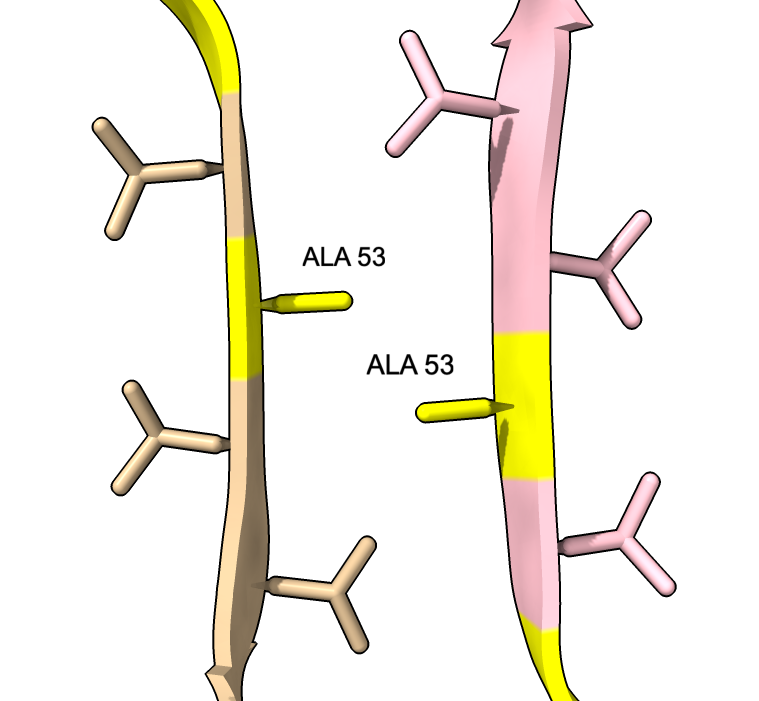
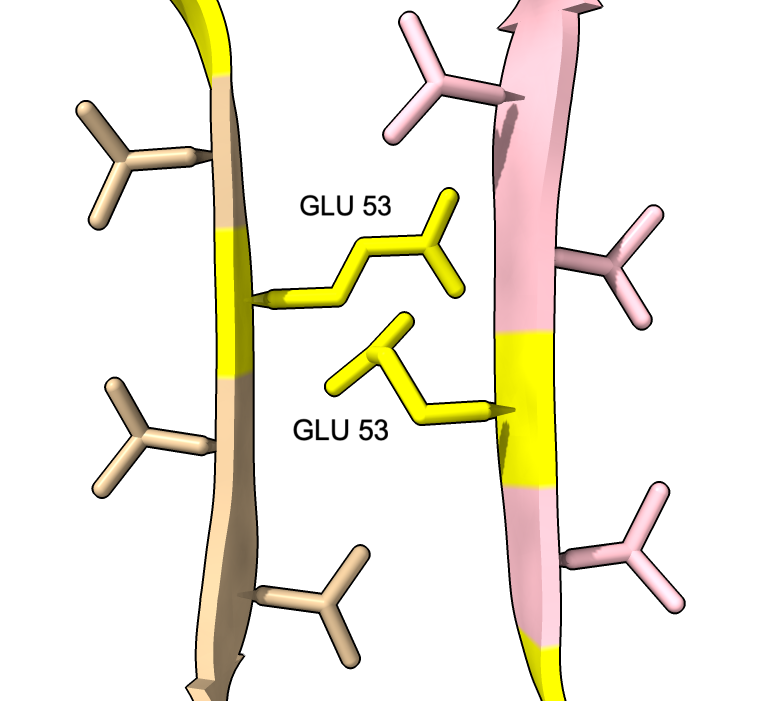
| color :46,50,51,53 yellow label :46,50,51,53 height 3 | Alanine 53 at proto-filament interface. | swapaa /I,G:53 GLU
label delete |
Details
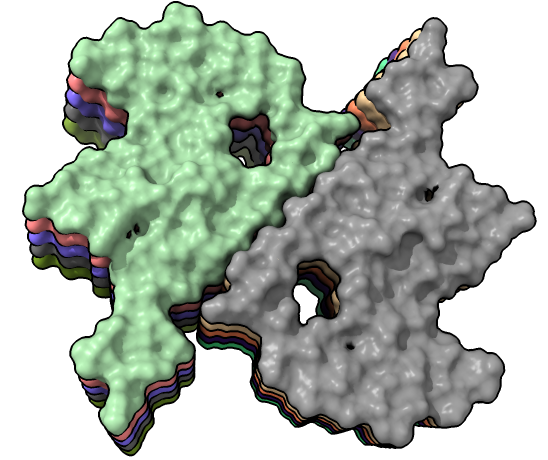
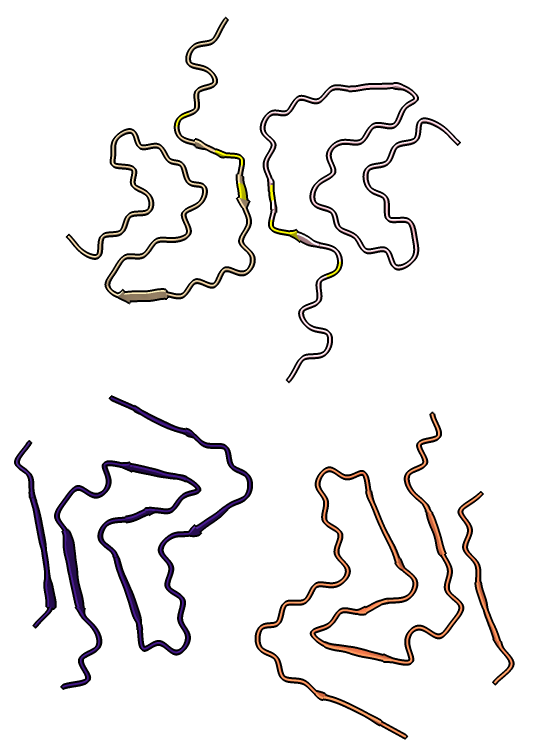
Four very different alpha-synuclein fibril structures have been published (PDB 6h6b, 6cu8, 6rt0, 6rtb, compared in Guerrero-Ferreira, bioRxiv preprint May 2019). Compare two of them.
| 
| |
| open 6rt0 show #2/J,E ribbon only hide atoms matchmaker #2/J to #1/I | tile | matchmaker #2/J to #1/I
delete #1 & ~/I,G delete #2 & ~/J,E morph #1,2 Press red circle button at right of morph slider to record movie. |
Details
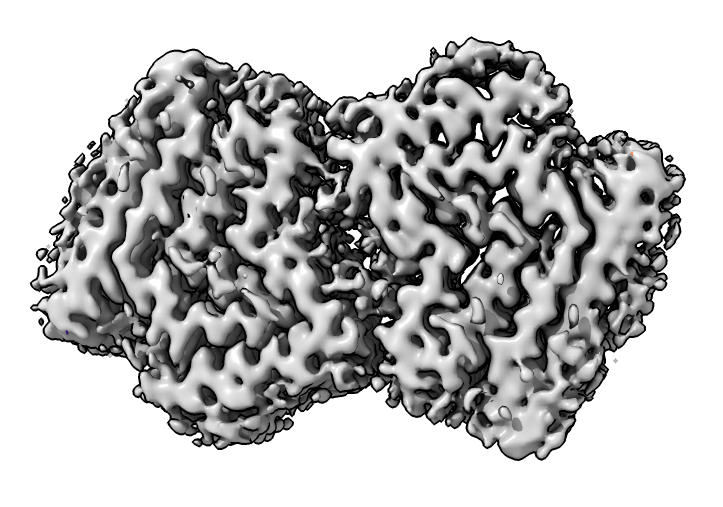
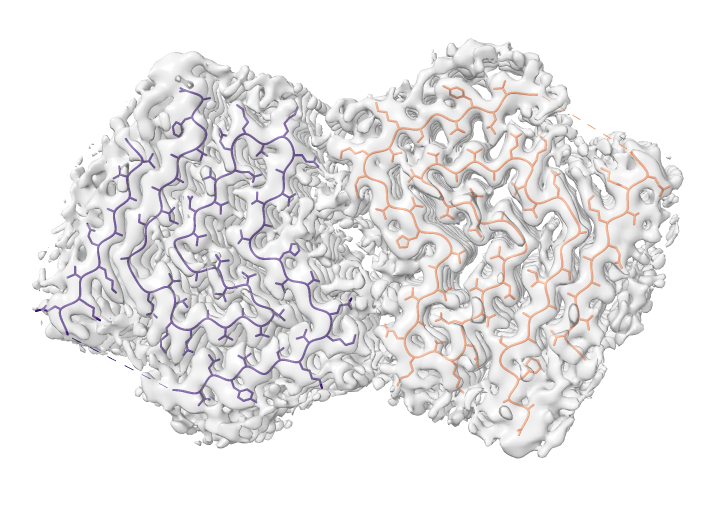
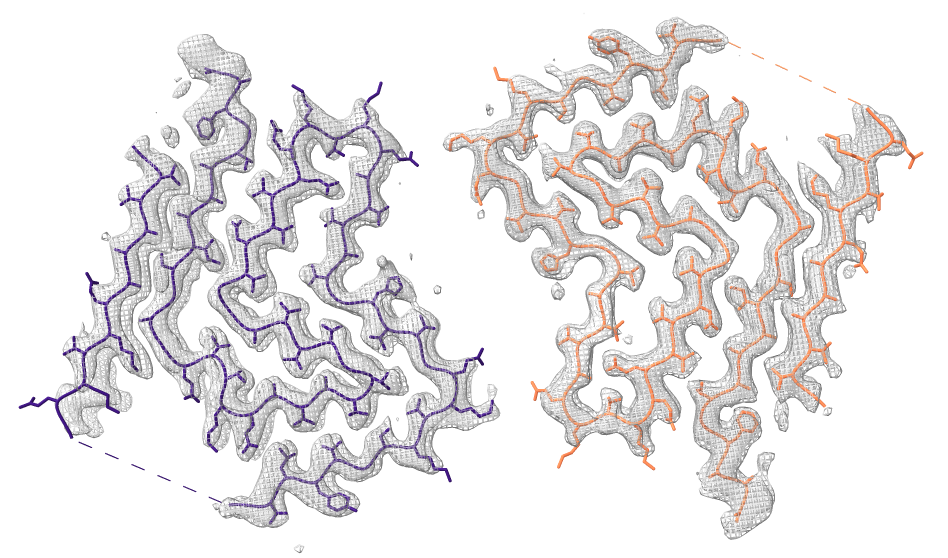
Look at cryoEM map EMDB 4994 used to determine atomic model PDB 6rt0.
| 
| 
|
| open 4994 from emdb | view initial #2
transparency #3 50 show #2 atoms,model | Use crop mouse mode to show thin slab of map.
volume #3 region 0,0,145,279,279,164 volume #3 level 0.01 volume #3 style mesh |
Details